


Executive Summary
- Immuno-oncology, also known as cancer immunotherapy, uses the body’s immune system to target and kill cancer cells.
- PD-1/PD-L1 inhibitors have emerged as effective forms of immunotherapy, but not all patients respond to these treatments.
- Many immuno-oncology targets are in different stages of clinical research with varying results.
- Effective precision oncology treatments will reach multiple targets and account for differences in each individual’s condition.
Over the past decade, immune checkpoint inhibitors targeting the PD1/PD-L1 axis have revolutionized cancer therapy, becoming foundational treatments across multiple tumor types. By blocking this inhibitory pathway, these agents restore cytotoxic T cell activity against malignant cells, producing durable clinical responses in cancers such as melanoma, non-small cell lung cancer, renal cell carcinoma, and microsatellite instability-high tumors. Since the landmark approval of pembrolizumab in 2014, PD-1/PD-L1 inhibitors have transformed treatment across more than 15 indications, establishing this pathway as a cornerstone of immuno-oncology and driving a global market of nearly US $50 billion annually.
Despite their transformative impact, however, PD-1/PD-L1 inhibitors are not universally effective. Data shows that even for patients with tumors highly positive for PD-L1, more than 50% might not respond to PD-1/PD-L1 blockade. Tumor heterogeneity, low mutational burden, impaired antigen presentation, and an immunosuppressive tumor microenvironment contribute to these limitations. “Cold” tumors with low T cell infiltration, such as pancreatic adenocarcinoma, glioblastoma, and microsatellite stable colorectal cancer, remain refractory to PD-1/PD-L1 blockade.
Among responders, acquired resistance frequently emerges through mechanisms including loss of major histocompatibility complex (MHC) expression, upregulation of alternative immune checkpoints, T cell exhaustion, and dynamic remodeling of the tumor microenvironment. Given these challenges, it’s critical that researchers identify and validate new targets that can function independently or in combination with existing therapies to improve patient outcomes.
Expanding precision oncology with more targets
The field of immuno-oncology remains at an early stage. While PD-1/PD-L1 inhibitors established a critical proof-of-concept for immune checkpoint modulation, their limitations underscore the need for broader mechanistic diversity. Given the complexity of the tumor microenvironment, no single target is likely to become a universal solution. Rather, effective therapeutic strategies will require complementary and multifaceted approaches.
In a recent study, the CAS team used a comprehensive approach including natural language processing (NLP) to analyze over 350,000 documents related to immuno-oncology in the CAS Content CollectionTM, the largest human-curated repository of scientific information. That analysis identified several emerging targets that seem to be paving the way for effective therapeutic strategies. In this article, we provide detailed and updated information on selected emerging therapeutic immuno-oncology targets (see Figure 1) and discuss where the field stands.
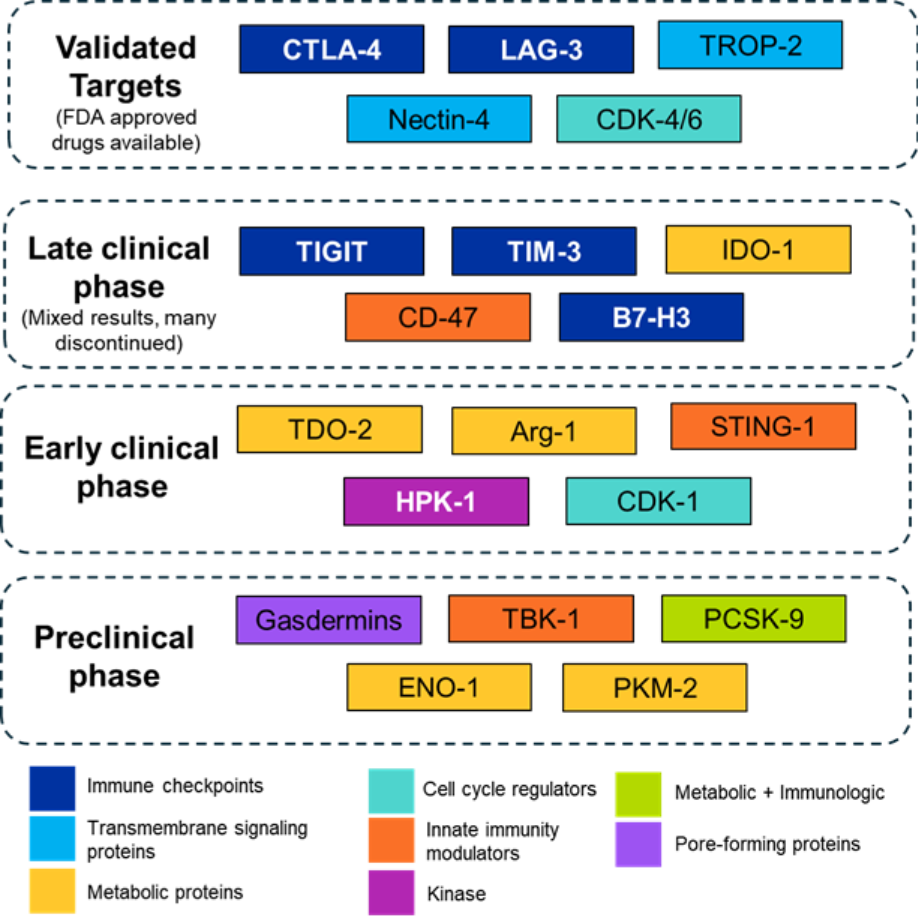
Figure 1: Overview of various emerging immune-oncological targets based on their clinical stage and mechanism. Source: CAS Content Collection.
Validated targets beyond PD-1/PD-L1 to overcome treatment resistance
The U.S. FDA has approved therapeutics against five non-PD-1/PD-L1 immuno-oncology targets so far (see Table 1). CTLA 4 (cytotoxic T-lymphocyte associated protein 4) pioneered immune checkpoint blockade by targeting T cell priming in lymphoid organs, garnering Nobel prizes for James P. Allison and Tasuku Honjo in 2018 in Physiology or Medicine. LAG 3 (lymphocyte activation gene 3) represents the first “next generation” checkpoint to translate into approval, validating the inhibition of exhausted T cell programs distinct from PD-1, with activity linked to co-expression biology and a more favorable safety profile, and reinforcing the importance of biomarker-informed development.
CDK4/6 (cyclin dependent kinase 4/6) inhibitors, originally approved as cell cycle agents, were subsequently shown to exert clinically relevant immunomodulatory effects illustrating how oncogene-targeted drugs can be repurposed to reshape antitumor immunity and sensitize historically immunologically “cold” tumors.
Finally, the antibody drug conjugate (ADC) targets TROP2 (trophoblast cell surface antigen 2) and nectin 4 demonstrate that cytotoxic antibody conjugates can function as immuno-oncology agents by coupling targeted tumor cell killing with immunogenic cell death, antigen release, and immune priming, particularly when paired with checkpoint blockade.
Collectively, these approvals confirm multiple viable paths to immune activation like checkpoint diversification, immune-modulating small molecules, and immunogenic cytotoxic platforms. These are expanding the therapeutic logic of cancer immunotherapy beyond PD-1 alone.
Table 1: U.S. FDA-approved drugs for immuno-oncological targets other than PD-1/PD-L1.
Immuno-oncology targets at pivotal validation stages
Multiple targets have advanced to Phase III trials, representing critical validation stages where preclinical promise and early clinical signals face definitive testing. This phase has produced mixed results offering valuable lessons for the field.
TIGIT (T cell immunoreceptor with Ig and ITIM domains) emerged as a promising checkpoint target based on robust preclinical data and encouraging Phase II results. Unfortunately, multiple TIGIT programs have failed thus far, including Phase III lung cancer trials and a melanoma Phase III study that was discontinued due to safety concerns.
However, Gilead Sciences and Arcus Biosciences are evaluating domvanalimab, a Fc-silenced anti-TIGIT, in Phase II trial for gastric cancer patients and are seeing positive results. Fc-silent antibodies are engineered antibodies that eliminate Fc gamma receptors (FcγR) binding in a bid to reduce undesirable immune responses and improve safety profiles. The data from this trial suggests that while first-generation TIGIT antibodies have struggled clinically, optimized approaches such as Fc silenced TIGIT blockade may still hold promise in selected tumor types.
Another emerging target, TIM-3 (T-cell immunoglobulin domain and mucin domain-3) has also shown mixed clinical results. Novartis discontinued the development of its anti-TIM-3 antibody sabatolimab after the primary endpoint was not met in a Phase II trial. GSK’s Phase III trial of cobolimab in combination with Jemperli and docetaxel also failed to improve overall survival in previously treated advanced non-small cell lung cancer.
However, this setback has not ended development, as cobolimab continues to be evaluated in early stage pediatric cancer studies and a mid-stage trial in advanced hepatocellular carcinoma. Several other bispecific antibodies targeting TIM-3 and PD-1 are showing promising results and remain of interest to researchers.
Similarly, CD47 targeting therapies have delivered mixed and often disappointing clinical results despite strong preclinical rationale. The early promise of Gilead’s magrolimab, particularly in TP53 mutant AML, failed to translate into survival benefit in Phase III and was compounded by anemia-related safety issues, leading to its discontinuation. Other anti-CD47 approaches (e.g., lemzoparlimab, bispecifics) have stalled or been halted for unclear reasons. Notably, evorpacept remains the lone CD47 program with encouraging safety and efficacy signals still advancing in solid tumors. Another modality, diphtheria toxin-based bivalent anti-human CD47 immunotoxin, has shown positive results in preclinical models.
B7-H3 (CD276) represents an unusual checkpoint target with controversial biology. Its receptor remains unknown, and whether it functions as a co-stimulatory or co-inhibitory molecule is debated. Despite this mechanistic uncertainty, B7-H3 has advanced to late-stage clinical development and various modalities like CAR-T cell therapies, bispecific antibodies, and ADCs. GSK’s risvutatug rezetecan is currently in Phase III trial. This is a novel investigational B7-H3-targeted ADC composed of a fully human anti-B7-H3 mAb linked to a topoisomerase inhibitor payload.
The next two to three years will be crucial in determining the fates of these candidates and whether any of these Phase III programs can join the approved list. The outcomes will further shape investment and development priorities for early-stage targets.
Precision oncology targets in early clinical stages
Several novel targets in Phase I/II development represent the next wave of innovation. These span metabolic checkpoints like TDO-2, arginase-1, innate immunity activators like STING-1, and kinases like MAP4K-1 and CDK-1.
We analyzed the CAS Content Collection using CAS IP Finder, powered by STNTM, to determine the number of documents for each of these targets over the past ten years (see Figure 2). By examining the yearly distribution of journal articles and patent families, we can see which targets are gaining the most interest with researchers. We also analyzed their patent-to-journal ratio to see which breakthroughs are closer to commercialization.
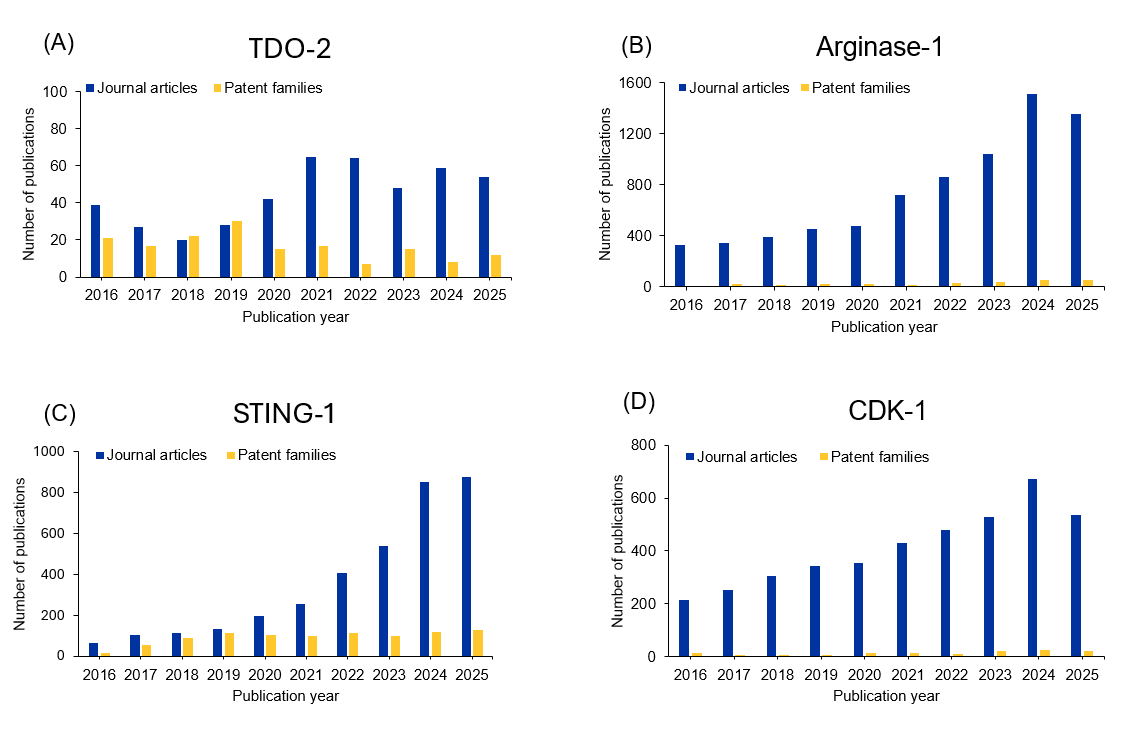
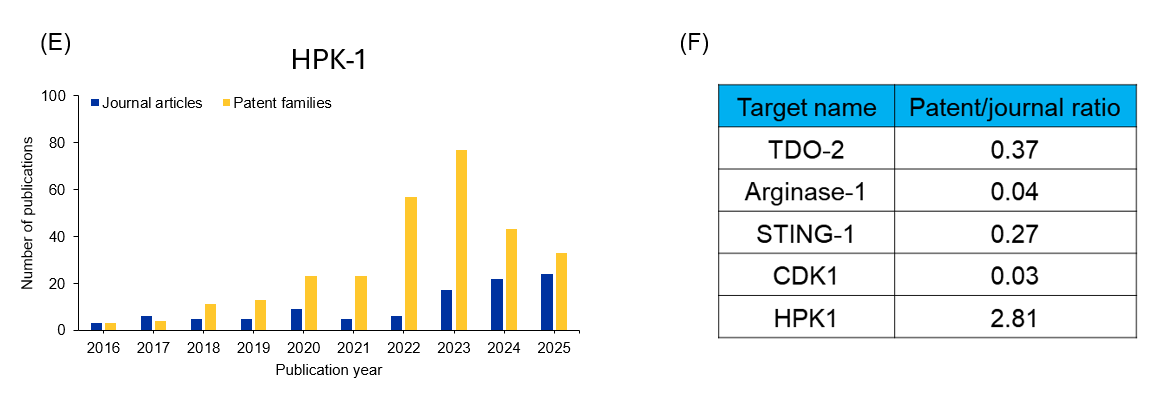
Figure 2: (A-E)Yearly publication trends for immune-oncological targets in early clinical stage; (F) shows patent-to-journal ratio. Source: CAS Content Collection.
The patent landscape analysis in Figure 2 used CAS IP Finder™ to identify filing patterns across jurisdictions. IP professionals monitoring competitive activity in this space can track new filings and identify whitespace opportunities using similar search strategies.
Five early-stage immuno-oncology targets shaping the next wave of cancer treatment
Failure in the field of metabolic checkpoints, exemplified by IDO-1, was mainly due to pathway redundancy as TDO-2 (tryptophan 2,3-dioxygenase) provides an alternative tryptophan-degrading mechanism. Given the IDO/TDO redundancy hypothesis, dual IDO/TDO inhibitors entered development. TDO-2 is expressed more selectively in tumors (particularly gliomas and hepatocellular carcinoma) compared to IDO-1's broader expression. Multiple Phase I/II trials of dual inhibitors are ongoing, proceeding cautiously with rigorous pharmacodynamic monitoring.
Arginase-1, expressed by myeloid-derived suppressor cells and tumor-associated macrophages, depletes L-arginine, disrupting T-cell function by blocking CD3ζ chain expression and limiting nitric oxide production. Unlike IDO, arginase-1 targets the myeloid compartment, offering mechanistic differentiation. The lead compound developed by Incyte Corporation, INCB001158, is an oral arginase inhibitor which completed Phase I/II trials. However, it showed limited antitumor activity when administered alone or in combination with pembrolizumab.
Following this result, multiple other trials of this compound were terminated, and the compound is no longer listed in the company portfolio, reflecting the end of active development. Unlike most other immunotherapeutics discussed in this article, INCB001158 is a small molecule inhibitor.
Beyond enzymatic inhibition, vaccination represents an emerging modality to target arginase-1. A first-in-human study of ARG1-derived peptide vaccines (combined with PD-L1 peptides) demonstrated favorable safety and robust ARG1 specific T cell responses in patients with myeloproliferative neoplasms, supporting further exploration of ARG1 vaccines in immuno-oncology.
The cGAS-STING (cyclic GMP-AMP synthase-stimulator of interferon genes) pathway detects cytosolic DNA, triggering type I interferon production and innate immune activation. Activating STING in the tumor microenvironment can convert immunologically "cold" tumors into "hot" tumors by recruiting and activating dendritic cells, promoting T-cell priming, and inducing inflammatory cytokines.
First-generation STING agonists, primarily cyclic dinucleotides, showed potent innate immune activation but were limited by poor cell permeability, rapid degradation, and inefficient access to STING, an endoplasmic reticulum localized transmembrane protein. To address these limitations, next-generation strategies include the development of orally bioavailable non-cyclic dinucleotides designed to penetrate cells and enable systemic dosing, alongside nanoparticle based delivery systems (lipid or polymeric) and targeted conjugates to enhance cytosolic delivery and tumor selectivity.
In parallel, RNA-based therapeutic approaches are emerging, including mRNA or self-amplifying RNA constructs encoding STING agonists or constitutively active STING variants, as well as synthetic RNAs that indirectly engage the STING pathway via upstream nucleic acid sensors. These advances aim to overcome delivery barriers, move beyond intratumoral administration, and improve the therapeutic window of STING pathway activation.
HPK1/MAP4K1 (hematopoietic progenitor kinase 1/mitogen-activated protein kinase 1) negatively regulates T-cell receptor (TCR) signaling by phosphorylating the 76 kDa leukocyte protein with an SH2 domain (SLP76), and other adaptor proteins, dampening T-cell activation. HPK1 inhibition enhances T-cell function, a cell-intrinsic mechanism distinct from blocking extracellular checkpoint receptors.
In recent years, significant progress has been achieved in the development of small-molecule inhibitors and proteolysis-targeting chimeras (PROTACs) targeting HPK1. Multiple small molecule inhibitors are reported, for example, FB-849, CFI-402411, GRC-54276, BGB-15025, BGB-26808, RGT-264, BB-3008, PRJ1–3024, and NDI-101150. These are being evaluated in Phase I/II trials either as standalone therapies or in combination with PD-1/PD-L1 checkpoint immunotherapy for various cancers.
For example, HDM2006 is a novel, orally active PROTAC targeting HPK1. Preclinical studies have shown an excellent profile for druggability, safety, and efficacy, and it has recently entered a Phase I trial. As seen in Figure 2, HPK1 has the highest patent-to-journal ratio of these early clinical targets, underscoring its potential to become an effective clinical treatment.
CDK-1 (cyclin dependent kinase-1) inhibition induces immunogenic cell death, releasing tumor antigens and damage-associated molecular patterns (DAMPs) that activate dendritic cells and prime anti-tumor T-cell responses. While CDK4/6 inhibitors have shown immunomodulatory effects and are approved, CDK1 is essential for all cell cycle phases and is more challenging to target safely. Multi-CDK inhibitors like dinaciclib (CDK1/2/5/9 inhibitor) are in clinical trials. However, selective CDK1 inhibitors mostly remain in the preclinical phase. Whether CDK1 inhibition will be tolerable and clinically successful in oncology remains to be determined.
We also analyzed the ligand landscape to understand the various modalities being explored for these five targets (See Figure 3).
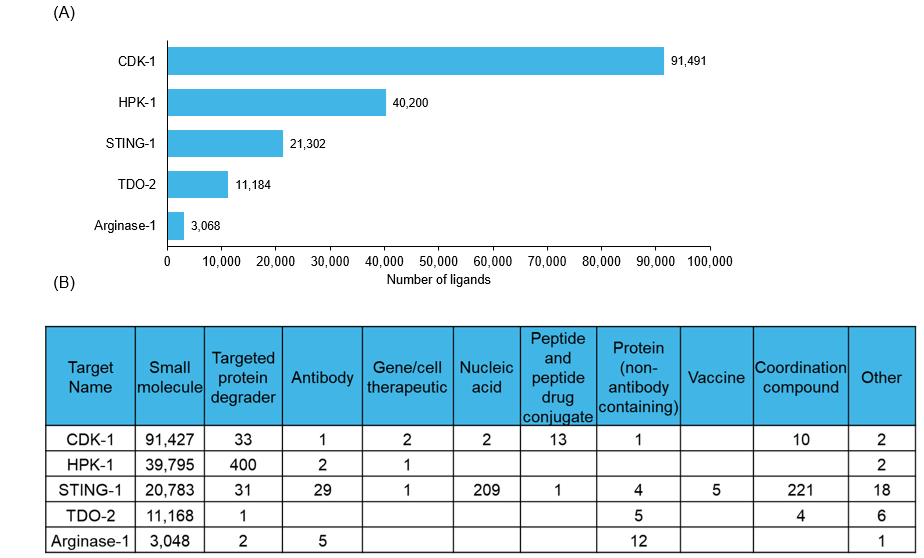
Figure 3: (A) The number of ligands and (B) ligand types reported against each target. Source: CAS Content Collection.
Figure 3A shows that CDK 1 dominates the ligand landscape by a wide margin (~91,000 ligands), followed by HPK 1 (~40,000) and STING 1 (~21,000), while TDO 2 and arginase-1 have substantially smaller ligand counts. Figure 3B further highlights clear modality preferences: CDK-1, HPK1, and TDO-2 are overwhelmingly explored via small molecules, whereas STING-1 exhibits the broadest modality diversity, with notable representation of nucleic acid therapies, vaccines, antibodies, and coordination compounds in addition to small molecules.
Immuno-oncology targets in the preclinical stage
Apart from these clinically established pathways, a diverse set of immuno-oncology targets is emerging in early preclinical stages. These modulate tumor immunity through inflammatory cell death, innate immune signaling, or metabolic reprogramming, offering complementary strategies to existing checkpoint-based therapies.
- Gasdermins, for example, are key executors of pyroptosis, a form of inflammatory programmed cell death that enhances tumor immunogenicity and immune cell recruitment. Therapeutic strategies aim to trigger gasdermin activation to convert immunologically “cold” tumors into inflamed, treatment responsive microenvironments. TBK 1 (tank-binding kinase 1) is an innate immune signaling kinase that regulates interferon responses and tumor cell resistance to inflammatory cytokines. Preclinical studies suggest that TBK 1 inhibition can sensitize tumor cells to immune-mediated killing and overcome resistance to immune checkpoint blockade.
- ENO 1 (α-enolase) is mainly a glycolytic enzyme with additional roles in immune modulation and tumor progression. Targeting ENO 1 reduces glycolysis and extracellular lactate accumulation. Additionally, it reprograms macrophage polarization and inhibits tumor growth and distant metastasis.
- PKM2 (pyruvate kinase M2) is a central regulator of aerobic glycolysis in cancer cells and influences immune cell differentiation and cytokine signaling. Preclinical studies show that targeting PKM2 aims to reprogram tumor metabolism and enhance antitumor immune activity within the tumor microenvironment.
- PCSK 9 (proprotein convertase subtilisin/kexin type 9) has emerged as a metabolic–immunologic target through its role in regulating MHC I degradation and tumor immune evasion. Inhibition of PCSK 9 in preclinical models enhances antigen presentation and improves responsiveness to T cell mediated immunotherapy.
These early clinical stage and preclinical stage candidates underscore the expanding landscape of immuno-oncology and highlight emerging opportunities to complement already established checkpoints through novel pathways.
Lessons learned and next steps for immuno-oncology targets
The mixed clinical outcomes observed across immuno-oncology targets reflect fundamental biological constraints rather than failures of trial design alone. The success of PD-1/PD-L1 blockade highlights the importance of targeting immune evasion mechanisms that are evolutionarily dominant and spatially confined to the tumor microenvironment. Conversely, many other proposed checkpoints lack comparable tumor microenvironment specificity, exert broader systemic immune regulation, or are not under strong selective pressure.
Tumor evolution further complicates therapeutic efficacy, as dominant suppressive pathways prevail until disrupted by therapy, after which adaptive resistance mechanisms may emerge under treatment-induced selective pressure. Together, these lessons highlight the need to move beyond simple checkpoint combinations toward mechanism-driven, biomarker-guided strategies tailored to tumor biology and patient context.
The selection of appropriate modalities is another critical aspect to consider, as the same target can respond differently to diverse modalities (e.g., B7-H3 shows varied responses to antibodies versus ADCs versus bispecific antibodies). Future progress in immuno-oncology will depend on identifying tumor specific, evolutionarily relevant, dynamically adapting therapeutic targets over time, expanding beyond classical checkpoints to orthogonal tumor microenvironment reprogramming approaches.
It’s important to remember that each regulatory approval expands the available therapeutic toolkit, each clinical failure provides instructive insights, and each early-stage target offers potential avenues to address unmet needs in patient populations that have yet to benefit from immunotherapy. Looking ahead, the coming decade is expected to be characterized by continued target diversification, maturation of biomarker-driven patient selection, and the integration of multiple therapeutic modalities into rationally designed combinations.




