


Strained spiro heterocycles are increasingly recognized as valuable three-dimensional bioisosteres in drug discovery. These rigid, three-dimensional molecular frameworks offer the pharmaceutical industry a potential solution to "escape from flatland" by providing scaffolds that can better complement complex biological binding sites compared to traditional flat aromatic compounds. Whereas the beneficial properties have been demonstrated extensively for spiro[3.3]heptanes, their lower homologues spiro[2.3]hexanes – a strained subset consisting of a three-membered ring fused to a four-membered ring through a shared spiro-carbon, remain largely unexplored due to synthetic challenges and limited biological data.
This is a missed opportunity, because beyond their rigid geometric structure, these strained spiro systems can induce enhanced target selectivity, improved solubility, increased metabolic stability, and reduced off-target effects. In addition, they also offer novel intellectual property advantages in previously inaccessible chemical spaces.
How can researchers better access these motifs to understand potential target-ligand interactions and predict which of these 3D candidates might be most promising for drug development? To answer this question, a team of scientists led by Professor Renzo Luisi at the University of Bari "A. Moro" in Italy developed a new synthetic approach to nine spiro[2.3]hexane analogues and used new AI-driven predictive analytics available in CAS BioFinder® to assess their potential as biologically active compounds. The project received funding support from the European Commission Horizon Europe Framework, Project SusPharma (Grant Agreement 101057430). The full results of this study have recently been disclosed as a research article in Angewandte Chemie International Edition.
This method is a practical way to accelerate research in this space by enabling rapid, literature-grounded in silico evaluation of ligand–target interactions and pharmacological potential, thereby helping to uncover a new class of therapeutically relevant molecules from this underexplored chemical territory.
Advantages of spiro[2.3]hexanes and synthesis challenges
The “escape from flatland” and “conformational restriction” concepts demonstrate that increasing the fraction of sp³-hybridized carbon centers in drug candidates is associated with higher success rates as compounds progress from discovery to clinical approval. Strained, sp³-rich spirocyclic scaffolds satisfy these criteria, making them valuable structural motifs in drug discovery and attractive to medicinal chemists.
Until now, medicinal chemists have devoted considerable attention to heteroatom-containing spiro[3.3]heptanes — two spirofused four-membered rings — and are exploring them as bioisosteric replacements for conventional, non-strained heterocycles such as piperazine or morpholine. These studies have shown that incorporating such strained, sp³-rich motifs can deliver compounds with improved physicochemical properties that progress into clinical trials.
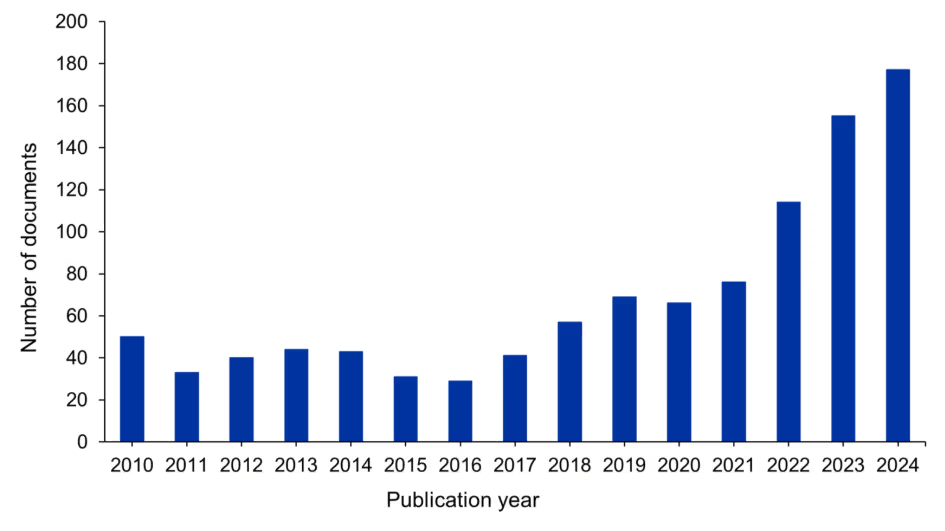
An analysis of publication data in CAS SciFinder® reveals the utility of the spiro[3.3]heptanes due to the presence of substantial number of reported synthetic derivatives and the large volume of patent applications and academic publications centered on this scaffold (see Figure 1). Although the lower homologue spiro[2.3]hexanes have attracted increasing academic attention over the past five years, demonstrated by a rising number of related publications shown in Figure 2, a systematic investigation of their bioisosteric potential remains elusive. This is despite their comparable physicochemical properties and reported biological activities, including inhibition of HDAC1/3 and HIPT1.
Researchers conducting similar searches across bioisosteric databases and materials literature can access training on structure-based queries and property filtering that demonstrates workflows for narrowing candidate materials.
Comprehensive CAS SciFinder data presented in Table 1 illustrates the prevalence of nine distinct spiro[2.3]hexane structural motifs reported in the literature. Notably, most heterocyclic analogues of spiro[2.3]hexanes remain underdeveloped, with limited academic reports and few or no patent applications identified.


Synthetic challenges associated with the efficient incorporation of such motifs, together with scarce chemical reactivity and stability data, have likely curtailed their widespread implementation in drug discovery.
Traditional syntheses of spiro[2.3]hexanes typically install the three-membered ring onto a preassembled four-membered scaffold via epoxidation, aziridination, or cyclopropanation [see Scheme 1A (i)]. These methods, however, often require harsh conditions, tolerate few functional groups, and lack modularity, as structural variation of the substituents on the three-membered ring necessitates resynthesis of the precursor.
A more flexible strategy has recently emerged based on intramolecular strainrelease reactions of bicyclo[1.1.0]butanes, enabling modular access to the spiro[2.3]hexane framework [see Scheme 1A (ii)]. Nonetheless, this approach still depends on reactive intermediates and is incompatible with the preparation of cyclopropane and oxetane-derived spiro analogues.
To address the aforementioned shortcomings and allow rapid access to a library of heteroatom-containing spiro[2.3]hexanes, the team designed novel sulfonium salts bearing four-membered rings, which can be transferred under mild conditions to a range of π-electrophiles (alkenes, carbonyls and imines) via a Johnson-Corey-Chaykovsky-type reaction to assemble nine different spiro[2.3]hexane motifs in a modular fashion, as shown in Scheme 1B.
A)
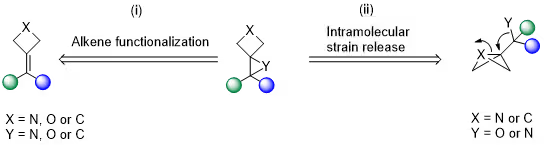
B)

Scheme 1. A) State of the art approaches for the synthesis of spiro[2.3]hexanes. B) A synthetic plan for the construction of spiro[2.3]hexane frameworks and their heteroatom-containing analogues employing four-membered ring-containing sulfonium salts as key precursors.
Synthesis of spiro[2.3]hexanes
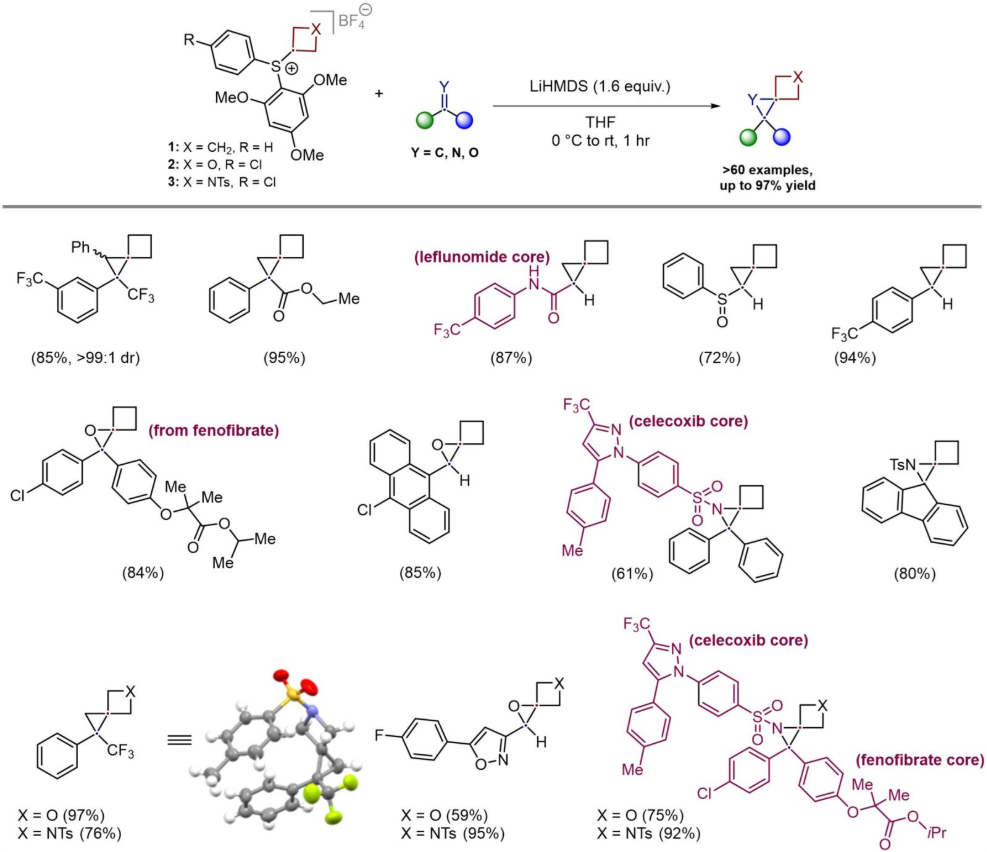
To realize the design plan shown in Scheme 1B, three novel sulfonium-based reagents bearing four-membered rings 1-3 (cyclobutane, oxetane, and azetidine) were developed. An aryl sulfide bearing the four-membered ring was found to be the key intermediate, which was oxidized to the sulfoxide, before being converted to the sulfonium salt by reaction with 1,3,5-trimethoxybenzene and triflic anhydride. This route was scalable to multigram-quantities and the reagents were found to be stable, free-flowing solids, meaning they are easy to handle in the laboratory, adding an important practicality aspect to the approach. Full details for the preparation can be found in the original article.
After optimization of the desired Johnson-Corey-Chaykovsky reaction, the practicality and modularity of this synthetic strategy was showcased by accessing over 60 substrates representing all nine spiro[2.3]hexane cores, fulfilling all predefined objectives. Some representative examples are shown in Scheme 2. For example, reaction with a range of electron-deficient alkenes, including styrenes, vinyl sulfoxides, acrylates, and acrylamides affords the desired spiro[2.3]hexanes and allows incorporation of pharmaceutically relevant cores, such as the leflunomide derivative. The differing reactivity of the sulfonium salt toward electron rich and electron deficient alkenes indicates that the reaction proceeds via nucleophilic attack by the ylide rather than carbene insertion. The corresponding epoxide derivatives are accessible by reaction with ketones and aldehydes, and this was amenable to the incorporation of active pharmaceutical ingredient fenofibrate. Last, the aziridine-bearing analogues can be obtained by reaction of the sulfonium salts 1-3 with imines. The strategy also enabled the installation of spiro[2.3]hexane motifs into complex imines, facilitated by the facile functionalization of the imine nitrogen. Accordingly, spiroaziridines incorporating the celecoxib core alone, as well as spiroaziridines bearing both celecoxib and fenofibrate cores, were successfully synthesized, demonstrating the capability of this method to deliver druglike molecules featuring spiro[2.3]hexane motifs (see Scheme 2). Notably, the reaction proceeded well regardless of the nature of the four-membered ring, allowing access to heteroatom-bearing spiro[2.3]hexane derivatives.
Systematic evaluation of bioisosteric potential of spiro[2.3]hexane analogues
The availability of all nine spiro[2.3]hexane analogues enables a systematic investigation of their bioisosteric potential, analogous to that of spiro[3.3]heptanes. To date, a thorough systematic characterization of these structural motifs has not been conducted.
To perform this evaluation, we developed a bioisostere identification strategy with a three-step workflow as presented in Scheme 3.
Step 1. Clustering-based approach to identify heterocycles with shared physicochemical properties:
First, an unsupervised learning approach was employed to compare all nine obtained (heteroatom-containing) spiro[2.3]hexane cores against a virtual database of over 70 commonly used drug discovery heterocycles to identify potential bioisosterism between spiro[2.3]hexanes and popular heterocycles in medicinal chemistry.
Thus, all structures were subjected to DFT optimization (ωB97XD3BJ/631++G(d,p)) to obtain energetically minimized three-dimensional conformations and their associated properties. One-dimensional (molecular weight, heteroatom count), two-dimensional (druglikeness, logP, topological polar surface area), and three-dimensional (dipole moment, plane of best fit, asphericity) molecular descriptors were selected and computed using RDKit to enable comparison of physicochemical properties and assessment of druglikeness.
To ensure robust statistical analysis, descriptors were manually inspected and excluded when log₁₀(VIF) ≥ 5. Dimensionality reduction via principal component analysis (PCA) and k-Medoids clustering enabled visualization of the high-dimensional dataset. PCA reduced the chemical space dimensionality and evaluated descriptor contributions, while k-Medoids clustering identified five distinct clusters (k = 5) based on Silhouette score analysis.
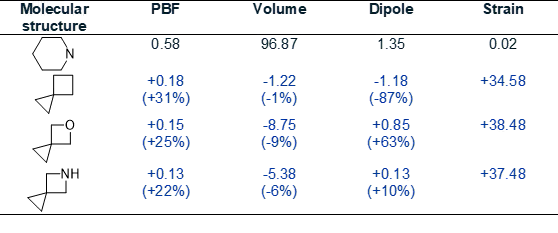
Eight of nine spiro[2.3]hexane analogues clustered together alongside pharmaceutically relevant heterocycles: isoxazole (found in leflunomide) and pyridine. Although in different clusters, piperidine showed spatial proximity to spiro[2.3]hexane cores. In 3D PCA space, piperidine and 5-azaspiro[2.3]hexane displayed average intra-cluster distances of 3.20 ± 0.76 and 2.28 ± 1.11, respectively, with an inter-cluster distance of only 1.69, confirming physicochemical similarity. Similarly, spiro[2.3]hexane showed an average intracluster distance of 3.59 ± 1.18 and a piperidine distance of 3.18. Compared to piperidine, spiro[2.3]hexane, 5-azaspiro[2.3]hexane, and 5-oxaspiro[2.3]hexane exhibited similar molecular volumes (crucial for binding site compatibility) and superior 3D drug-likeness indicators (PBF). Notably, 5-azaspiro[2.3]hexane showed comparable dipole moment, but increased strain energy. The clustering proximity and molecular descriptor similarities support 5-azaspiro[2.3]hexane as a promising strained piperidine bioisostere (see Table 2).
Step 2. AI-driven prediction of target–ligand interactions to refine candidate selection for a specific biological target:
Having established in-silico the potential similarity with piperidine, this hypothesis was tested on a real-life example. Pethidine, a piperidine-containing μ-opioid receptor agonist, was selected as a test case due to its structural compatibility with the current methodology and clinical need for safer alternatives (given toxic norpethidine metabolite formation) (see Figure 3a). To identify the most likely analogues to be active against the μ-opioid receptor and prioritize candidates for in vitro testing, we turned to AI-enhanced predictive analytics. CAS BioFinder was selected for this purpose. This platform enabled rapid, data driven in-silico prediction of pharmacological activity by modeling protein–ligand interactions using curated chemical–biological relationships extracted from scientific literature. An iterative predictive analytics workflow was applied to evaluate pethidine analogues containing the 5-azaspiro[2.3]hexane core against the human μ-opioid receptor (see Figure 3b).
Initial screening (pAct and confidence scores; Table 3) identified analogues 4 and 5 as promising, with predicted pAct values of 6.54 and 5.92, respectively. The spiro[2.3]hexane and 5-oxaspiro[2.3]hexane analogues (6 and 7), identified as less promising by clustering, showed no predicted activity. Guided by these results, further 5-azaspiro[2.3]hexane derivatives were explored by modulating the ester side chain. Substituting the ethyl ester with methyl (8) or isopropyl (9) groups caused only slight decreases in predicted pAct (6.50 and 6.48). Interestingly, introducing an azetidine ester (10) substantially increased the predicted activity (pAct 7.22), whereas the oxetane ester (11) showed no predicted activity against the evaluated targets.
![Structures of pethidine and the uploaded spiro[2.3]hexane derivatives tested against it.](https://cdn.prod.website-files.com/650867962272bf8f15c1034b/69cd636d726d7a8ccab9336e_download.avif)
![Predicted activity scores of spiro[2.3]hexane derivatives at the mu-type opioid receptor.](https://cdn.prod.website-files.com/650867962272bf8f15c1034b/69cd63aede6a3f875713e763_download.avif)
Step 3. In vitro validation of prioritized ligands demonstrating high interaction probability with the selected target:
To experimentally evaluate the predictive analytics approach, the three ligands with the highest predicted activity (pAct) against the μ-opioid receptor (compounds 4, 8, and 10) were selected for in vitro testing, together with spiro[2.3]hexane 6 as a negative control. All these compounds were synthesized using the developed methodology and were assessed using a label-free binding assay (EnSpire platform, PerkinElmer) employing SHSY5Y neuroblastoma cells expressing μ-opioid receptors. Serial dilutions (0.03–150 μM) were analyzed via optical biosensors detecting refractive index changes, with DAMGO, a known μ-opioid receptor agonist, used as a positive control.
Dose–response analysis revealed micromolar binding activity (10–39 μM) for all tested compounds, confirming μ-opioid receptor engagement (see Figure 4). The observed binding of the predicted active compounds (4, 8, and 10) is consistent with the in silico predictions and validates the suitability of the 5-azaspiro[2.3]hexane core as a substituted piperidine bioisostere. Analogue 10, for which the highest pAct was predicted on CAS BioFinder, showed the best binding activity (10 μM).
While unsupervised learning enabled identification of generally suitable bioisosteric scaffolds, AI-driven target prediction effectively prioritized candidates for experimental validation. Notably, compound 6, which was predicted to be inactive despite structural proximity to piperidine in the clustering analysis, also displayed measurable binding activity, highlighting current limitations of predictive models when applied to scaffolds with limited representation in available training data.
![Four spiro[2.3]hexane derivatives with measured EC50 values from 10 to 39 micromolar.](https://cdn.prod.website-files.com/650867962272bf8f15c1034b/69cd63f5b5c481c802e57b91_download.avif)
New approaches for bioisostere identification in drug discovery
A simple, general, and functional group tolerant strategy was developed for accessing previously underexplored spiro[2.3]hexane analogues using three novel sulfonium salt reagents. This synthetic platform was shown to be broadly applicable, with electron deficient alkenes, carbonyl compounds (ketones and aldehydes), and imines serving as effective reaction partners for the formation of spirocyclic cyclopropanes, epoxides, and aziridines, respectively. In total, over 60 examples were reported, highlighting the versatility and robustness of the methodology.
Beyond synthetic development, the bioisosteric potential of spiro[2.3]hexanes was systematically evaluated using an integrated in silico workflow combining unsupervised learning and AI-enhanced predictive analytics. Application of CAS BioFinder enabled data-driven prediction of protein–ligand interactions based on curated chemical–biological relationships extracted from scientific literature. By narrowing the candidate list from 66 compounds to four likely candidates in just a few minutes, CAS BioFinder saved at least $15,000 on external testing for this single project and accelerated the overall project timeline to eight months from initial project design to biological validation. Similar projects often take years without CAS BioFinder.
This analysis identified 5-azaspiro[2.3]hexane as a promising piperidine bioisostere based on its predicted structural and interaction features. Using pethidine, a piperidine-containing μ-opioid receptor agonist employed in obstetric analgesia, as a model system, this hypothesis was subsequently validated through in vitro binding studies.
Overall, this work demonstrates how modern AI-enabled predictive tools such as CAS BioFinder can complement synthetic chemistry by guiding target-focused compound selection and accelerating the timeline between discovery of novel chemical space and biological evaluation in vitro. With transparent and well-founded predictions from CAS BioFinder, synthetic chemists can more easily identify targets for completely novel molecules that would not have been designed by rational drug design in the first place. Starting in vitro testing with a shorter, pre-screened candidate list reduces the risks and costs for in vitro testing through pre-validation and focus.
It is anticipated that the sulfonium salt reagents described herein will find broader application in organic synthesis, and that this modular route to spiro[2.3]hexanes, combined with predictive analytics, will facilitate further exploration of these motifs as valuable cores in medicinal chemistry.
---
Watch the recording here of a webinar where Dr. Natho walked through a practical, AI-powered workflow that makes the process of finding the right molecular scaffold faster and more efficient. They show how a flexible new synthesis method combined with AI-driven predictive analytics can quickly identify the most promising candidates before committing to expensive lab testing.
---
P.Natho, A.Vicenti, F.Mastrolorito, F.De Franco, L.Walsh-Benn, M.Colella, E.Mesto, E.Schingaro, O.Nicolotti, A.Gioiello, R.Luisi, Angew. Chem. Int. Ed. 2026, 65, e21633. https://doi.org/10.1002/anie.202521633




