
Biotechnology
From batteries to biosensors: Conductive polymers make the jump to commercial applications
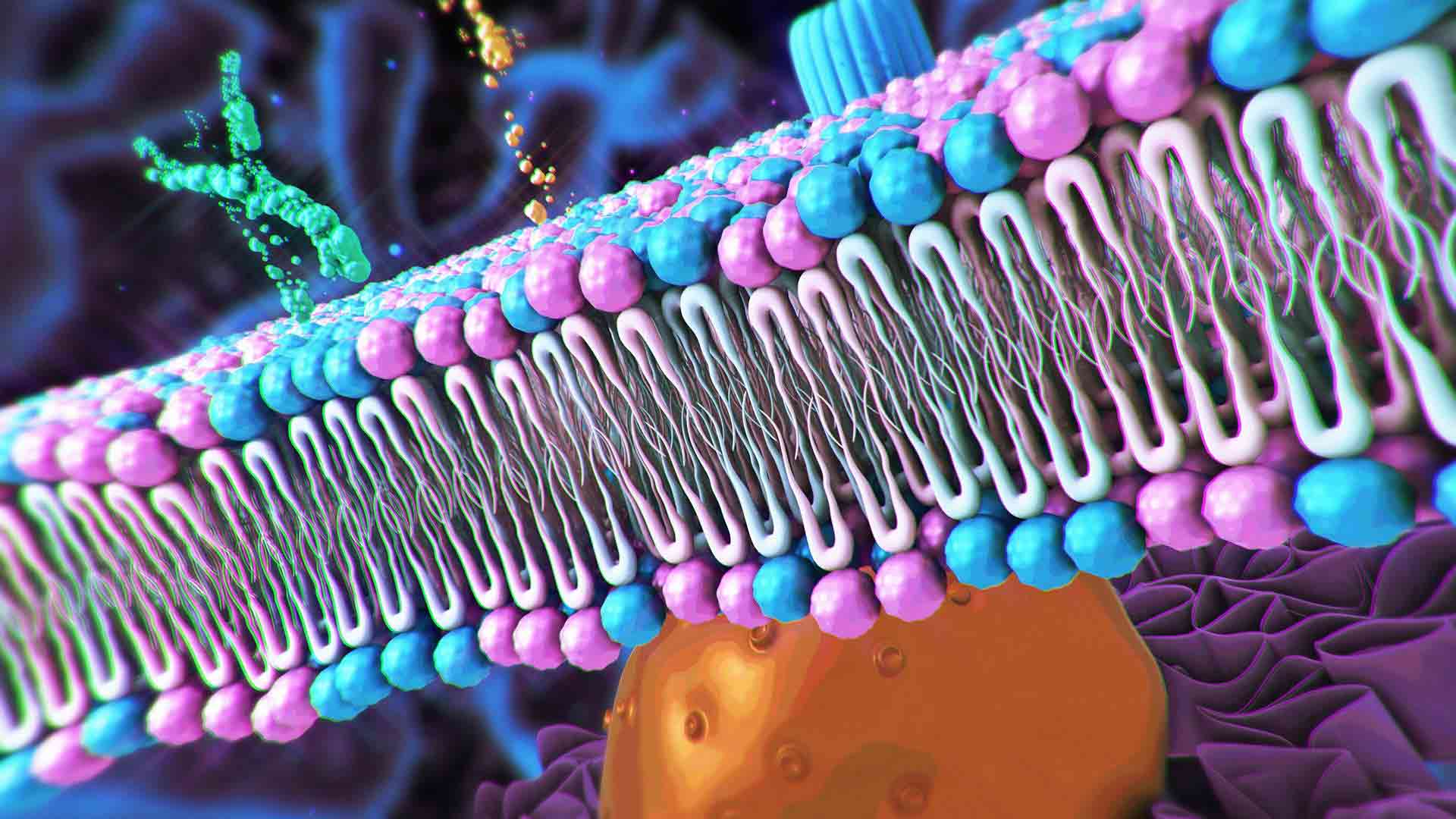
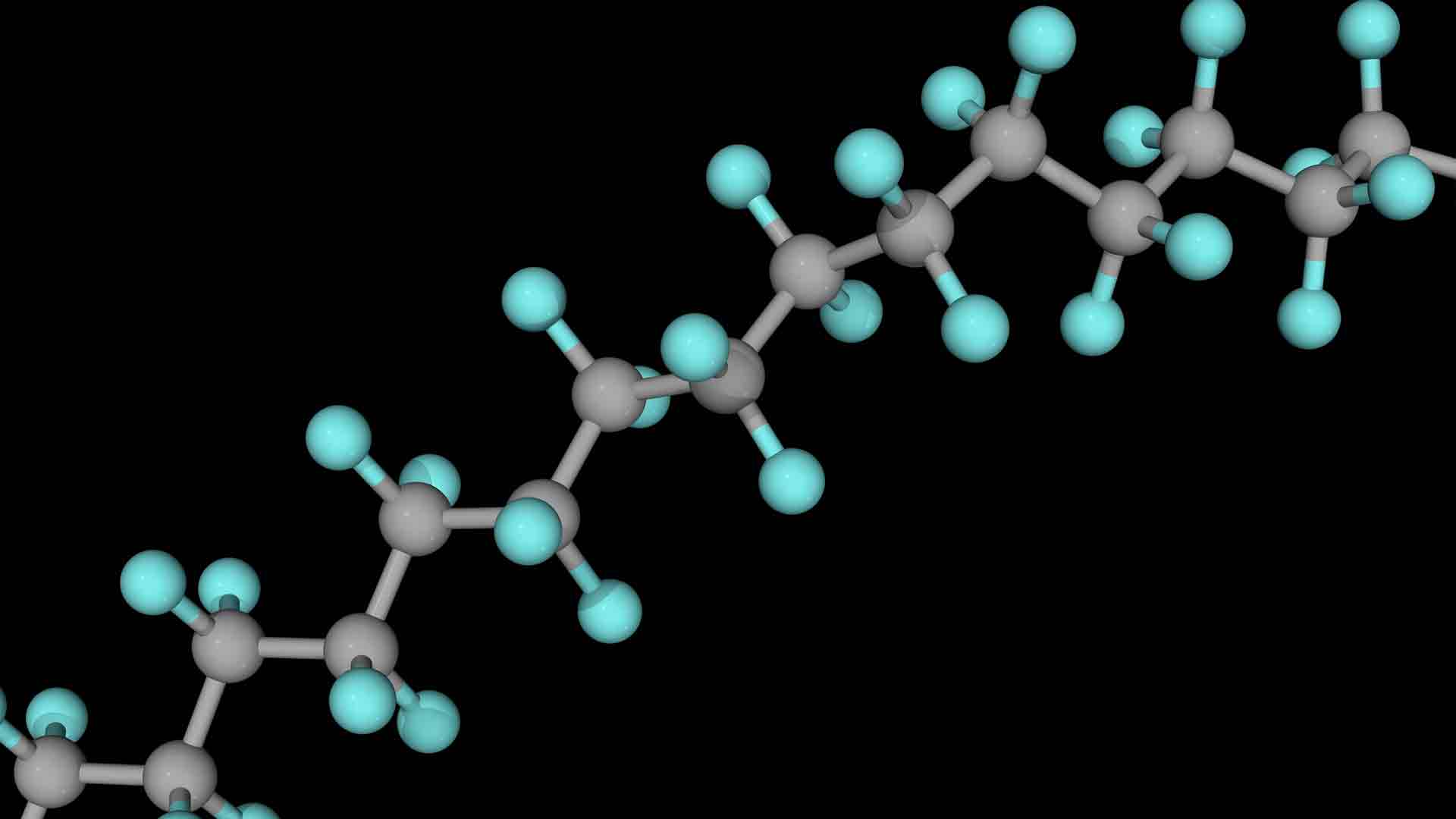
Revolutionary materials that conduct electricity like metals while bending like plastics are transforming everything from batteries to brain implants. Discover how conductive polymers went from Nobel Prize discovery to cutting-edge biomedical breakthroughs.
Read the reportRead the articleDownload the summarySee the infographicRead the publicationRead the recapWatch the video













.avif)