

Peptide therapeutics have emerged as a promising modality in human medicine over the past several decades, owing to their high specificity, favorable safety profiles, and expanding chemical design space. We see this most recently in the explosive growth of GLP-1 receptor agonists like semaglutide. Among all FDA approved peptide therapeutics, about 25% belong to the category of cyclic peptides. Cyclic peptides are a class of polypeptides in which the amino acid chain is covalently closed to form a ring structure through various cyclization strategies. This topology distinguishes them from linear peptides and imparts enhanced conformational rigidity, proteolytic stability, and better binding affinity.
Typically, cyclic peptides occupy an intermediate molecular weight range of ~500–3000 Da, situating them between small molecules and large biologics in size and complexity. Their cyclic architecture imparts several advantages, including enhanced resistance to enzymatic degradation, improved target binding, and, crucially, the potential for oral bioavailability, a long-standing hurdle in peptide-based therapeutics. These structurally constrained compounds occupy a unique chemical space, combining the high specificity and affinity of large biologics with the favorable pharmacokinetic properties of smaller molecules.
The global cyclic peptide market is experiencing steady growth, driven by their unique structural advantages compared to linear peptides. The market is projected to reach nearly USD $3.6 billion in 2026 with a CAGR of 6.5-6.7%, leading to about USD $5.3 billion by 2032. Biopharmaceutical applications dominate, particularly in oncology, autoimmune disorders, and infectious diseases, supported by advancements in solid-phase synthesis, display screening platforms (like mRNA display and phage display), and computational design.
Key players in the cyclic peptides market include Bicycle Therapeutics, Merck, Bachem, and Apellis Pharmaceuticals, while emerging uses in diagnostics, environmental protection, and biosensing are expanding the market footprint. This growth reflects cyclic peptides’ role as a cornerstone modality in next-generation biologics development.
We explored data from the CAS Content CollectionTM , the largest human-curated collection of published scientific information, to provide a comprehensive overview of cyclic peptides with a focus on their therapeutic potential. Our findings indicate that the oral route of delivery is gaining increasing attention, as reflected by a steady rise in publications over the past several years.
Beyond delivery, we also examined how specific properties like peptide types, cyclization types, and certain peptide modifications co-occur with therapeutic areas, potential molecular targets, and administration routes. Together, these insights provide a comprehensive view of emerging trends that are shaping the future development of cyclic peptide therapeutics.
Trends in cyclic peptide research from CAS data
To map the publication landscape, we accessed data from the CAS Content Collection via CAS SciFinder®https://www.cas.org/solutions/cas-scifinder-discovery-platform and CAS STNext® tools and performed a quantitative analysis. This revealed a steady growth trajectory from 2006 to 2025, with journal publications consistently dominating the research output (see Figure 1). Journal articles account for approximately 66% of total publications, while patents represent 34% , indicating a strong emphasis on fundamental research alongside commercial applications.

Year-wise trends show a notable acceleration in publication activity beginning around 2015, with peak output observed during 2021-2024. Notably, in 2023 and 2024, patent publications surpassed journal articles, suggesting an increasing focus on commercialization and applied research in recent years.
We then examined CAS section data, resulting from robust indexing by CAS analysts, to understand the major areas of innovation relating to cyclic peptides (see Figure 2). Overall, pharmaceuticals consistently dominate the landscape, showing a sharp rise from 2022 to a peak in 2024 (>2-fold increase), reflecting strong translational interest in cyclic peptides for therapeutic development.
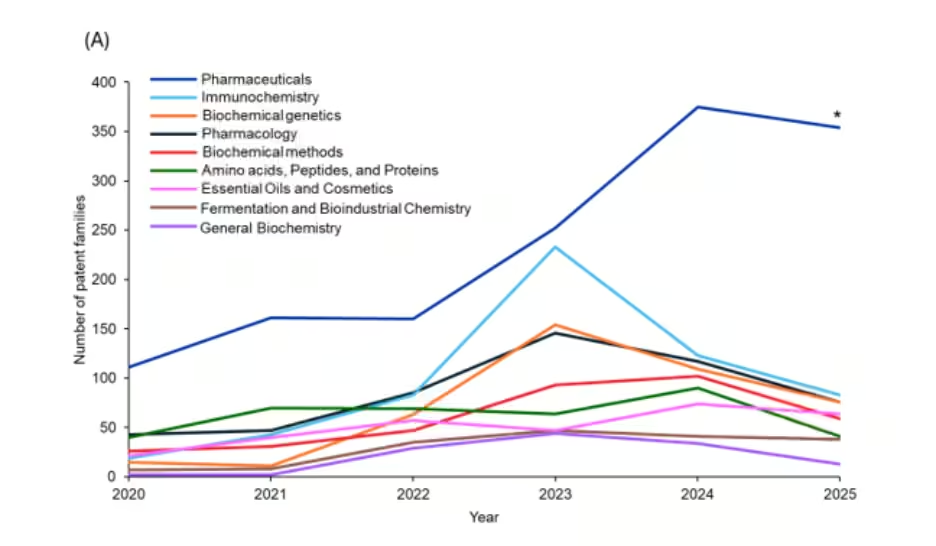
Figure 2: Yearly trends of patents related to cyclic peptides based on their respective CAS sections. *Data for 2025 is partial through August. Source: CAS Content Collection.
Immunochemistry, biochemical genetics, pharmacology, and biochemical methods also show notable surges, suggesting growing activity in peptide-based immunomodulators and production methodologies. Together, these trends indicate that the growth in cyclic peptide patents is driven not only by therapeutic applications but also by their expanding use in biochemical tools, platform technologies, and industrial biotechnology.
Cyclic peptide as therapeutics
To investigate emerging trends in cyclic peptides therapeutics, we conducted a detailed analysis of substance data from 2020-2025 in the CAS REGISTRY®. We included in our analysis substances indexed with the following CAS roles: therapeutic (THU), pharmacological (PAC), or pharmacokinetic (PKT). Their corresponding SMILES representations were processed using RDKit for structural evaluation.
By applying this workflow, we identified 46,574 cyclic peptides. Classification was based on the following criteria: (1) The molecule contains at least one ring (has_any_ring = True); (2) The molecule possesses ≥ 2 amide bonds overall (len(amide_bidx) ≥ 2); (3) At least one ring incorporates ≥ 2 backbone-like amide bonds, with both termini adjacent to α‑carbons. A peptide was considered a cyclic peptide only if all these three conditions were true.
We then analyzed the major therapeutic areas mentioned in our dataset and looked at the distribution of these cyclic peptides across them. The analysis shows a clear dominance in oncology (see Figure 3).
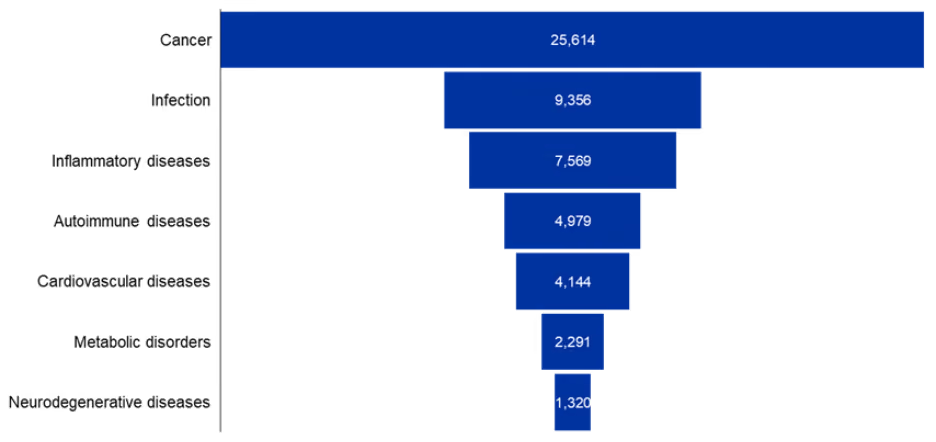
Figure 3: Major therapeutics areas in cyclic peptides research. Each bar shows the number of cyclic peptides associated with a particular therapeutic area. Data for the period 2020-2025. Source: CAS Content Collection.
Infectious and inflammatory diseases represent the next major clusters. Autoimmune and cardiovascular diseases also feature prominently, though with substantially fewer publications compared to cancer. Metabolic and neurodegenerative disorders represent smaller but emerging areas of exploration. Overall, these trends highlight that while cyclic peptides are being investigated across a broad therapeutic landscape, cancer and infectious diseases remain the primary drivers of research activity.
Having identified more than 46,000 cyclic peptides, our next step was to analyze these identified peptides using RDKit to determine the types of cyclic peptides (based on the number of amino acids they contain), the type of cyclization present in a given peptide, and which, if any, modifications were present on these peptides.
[H3]: Types of cyclic peptides
Cyclic peptides represent a structurally diverse class of biomolecules that can be categorized according to their source, the nature of the bonds forming the ring, and the number of amino acid residues involved. From a source perspective, they may be either naturally occurring, produced by microorganisms, plants, or marine organisms, or synthetically generated through chemical synthesis for tailored pharmacological applications.
Based on the type of bond, cyclic peptides can be divided into homodetic, isopeptide, and depsipeptide classes. Homodetic cyclic peptides, such as cyclosporine A, are characterized by rings composed exclusively of standard peptide bonds between the α-carboxyl group of one residue and the α-amino group of another.
Isopeptide cyclic peptides, exemplified by microcystin and bacitracin, contain at least one non-α amide linkage, often involving side chains, which impart structural diversity and unique biological activity.
Depsipeptides, including aureobasidin A, kahalalide F, and didemnin B, feature at least one ester (lactone) linkage in place of an amide bond, frequently formed between the C-terminal carboxyl group and the hydroxyl side chain of serine or threonine residues, and are notable for their potent pharmacological properties.
Classification by ring size further highlights the functional diversity of cyclic peptides. Cyclic dipeptides, or diketopiperazines, are the simplest members, typically rigid and resistant to proteolysis. Tripeptides, slightly larger and more conformationally flexible, have been reported to exhibit antioxidant and anti-inflammatory properties. Tetrapeptides, with reduced ring strain, display enhanced stability and are widely studied for receptor modulation and pharmacological activity. Pentapeptides strike a balance between conformational diversity and stability, making them attractive scaffolds for drug design with improved bioavailability.
In general, tetra- and pentapeptides are considered the threshold for reliable stabilization, as smaller rings such as di- and tripeptides may suffer from ring strain, while larger macrocyclic structures gain conformational stability and functional versatility. Macrocyclic peptides, as well as bicyclic and polycyclic architectures, exhibit exceptional stability and highly specific binding properties, exemplified by molecules such as vancomycin, daptomycin, and defensins. For reliable stabilization, smaller rings such as di- and tripeptides may suffer from ring strain, while larger macrocyclic structures gain conformational stability and functional versatility. Macrocyclic peptides, as well as bicyclic and polycyclic architectures, exhibit exceptional stability and highly specific binding properties, exemplified by molecules such as vancomycin, daptomycin, and defensins.
The term “macrocyclic peptide” has been vaguely defined in the literature, and there is no known stringent cut-off on the number of amino acids in a macrocyclic peptide. Here, based on previously published reports and opinions of experts in the field, we have considered any cyclic peptide with six or more amino acids as a macrocyclic peptide. Bicyclic or polycyclic peptides have complex structures with two (bi-) or more (poly-) cyclic rings. Collectively, these classifications underscore the structural and functional richness of cyclic peptides, which continue to serve as valuable templates in drug discovery, chemical biology, and therapeutic development.
[H3]: Types of cyclization
Cyclic peptides exhibit remarkable structural diversity, and the mode of cyclization strongly influences their stability and biological properties. Head-to-tail cyclization is the most prevalent, forming a closed ring between the N-terminal amine and C-terminal carboxyl group (see Figure 4). This orientation eliminates free termini, conferring resistance to exopeptidase degradation and often enhancing membrane permeability and intracellular delivery.
Head-to-side and side-to-tail cyclizations introduce alternative ring closures by linking termini to reactive side chains, which can fine-tune conformational rigidity and surface exposure of functional groups. Side-to-side cyclization, such as disulfide bond formation between cysteine residues, stabilizes secondary structures and allows reversible redox control, making it useful in mimicking natural peptide hormones and toxins.
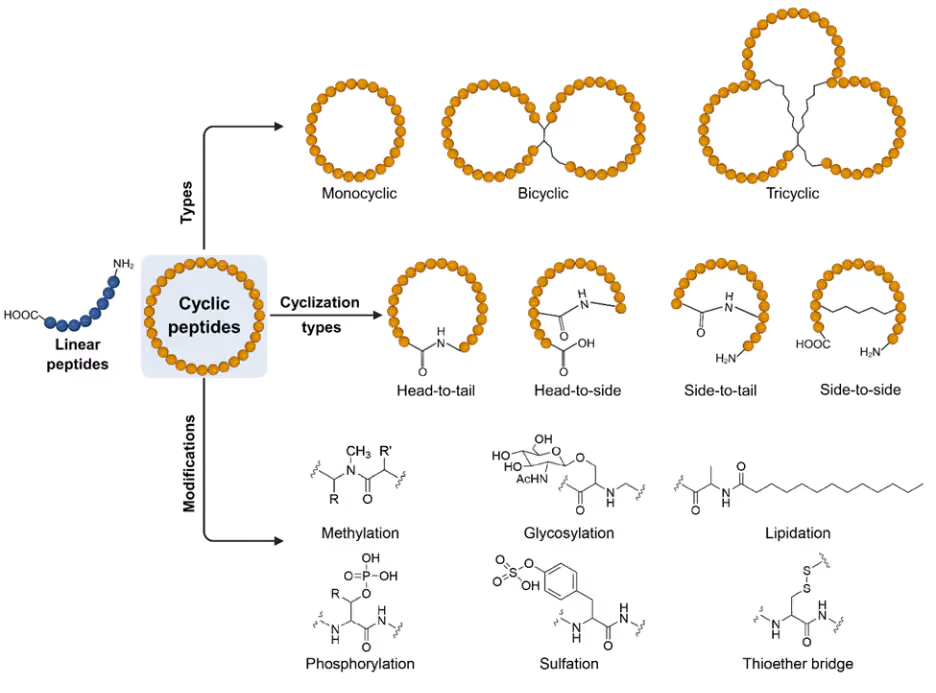
Figure 4: Schematic representation of the types of cyclic peptides, their cyclization types and a few common modifications. Figure created partially using www.BioRender.com.
Beyond these canonical strategies, mixed mode cyclization combines multiple linkages to create hybrid architecture with enhanced complexity. Such designs can lock peptides into highly defined conformations, improving receptor selectivity, bioavailability, and thermal stability. By carefully choosing the cyclization orientation, chemists can modulate peptide solubility, resistance to enzymatic breakdown, and overall pharmacokinetic behavior.
As noted, we analyzed cyclic peptides with CAS roles THU, PAC, and PKT from the CAS Content Collection using RDKit to classify them based on their type and cyclization type (see Figure 5). For this, we selected the ring with the highest number of amide bonds (most likely the main macrocycle). We then counted all amide bonds in that ring (ignoring the α-carbon filter for size naming). Lastly, we applied the following classification: if the peptide contains 2 amides → cyclic dipeptide; 3 amides → cyclic tripeptide; 4 amides → cyclic tetrapeptide, 5 amides → cyclic pentapeptide; and ≥ 6 amides → macrocyclic peptide. If there were more than one amide-containing rings, then the peptides were classified as bicyclic or polycyclic.
To determine the cyclization type, carbonyl carbon and amide nitrogen atoms were identified and checked for adjacency. If amide N was adjacent to an α-carbon, it was marked as head (backbone). If carbonyl C was adjacent to an α-carbon, it was marked as tail (backbone). The cyclization type was determined based on whether the two ends involved both as backbone (head-to-tail), one N backbone and other as C side-chain (head-to-side), one N side-chain and other as C backbone (side-to-tail), and both ends as side-chains (side-to-side).
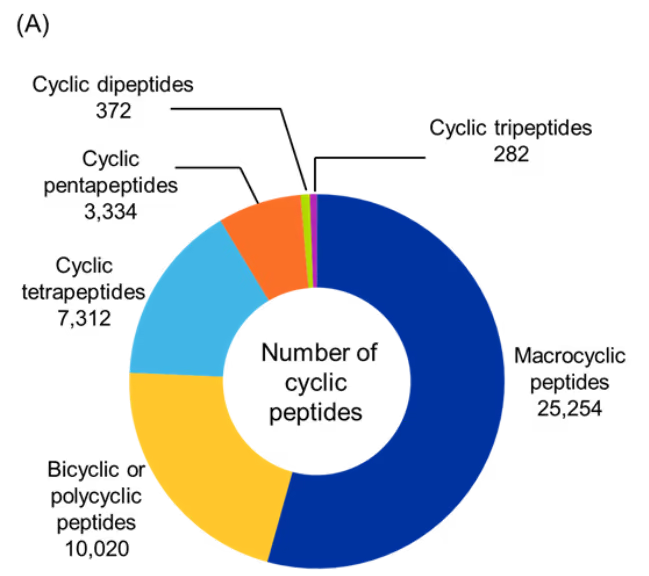
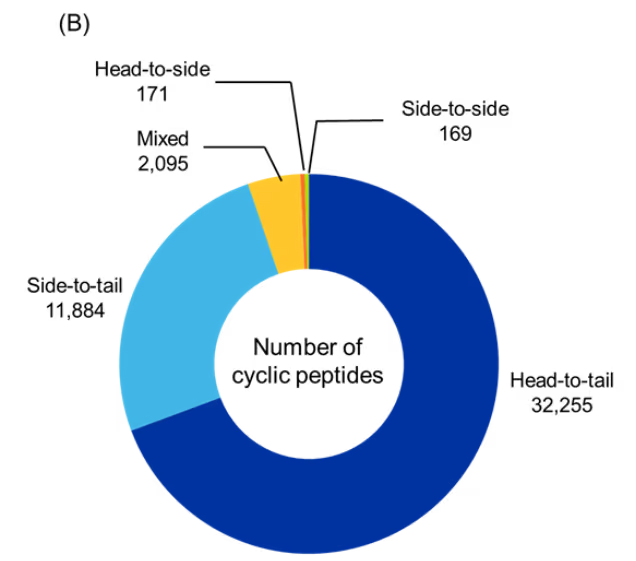
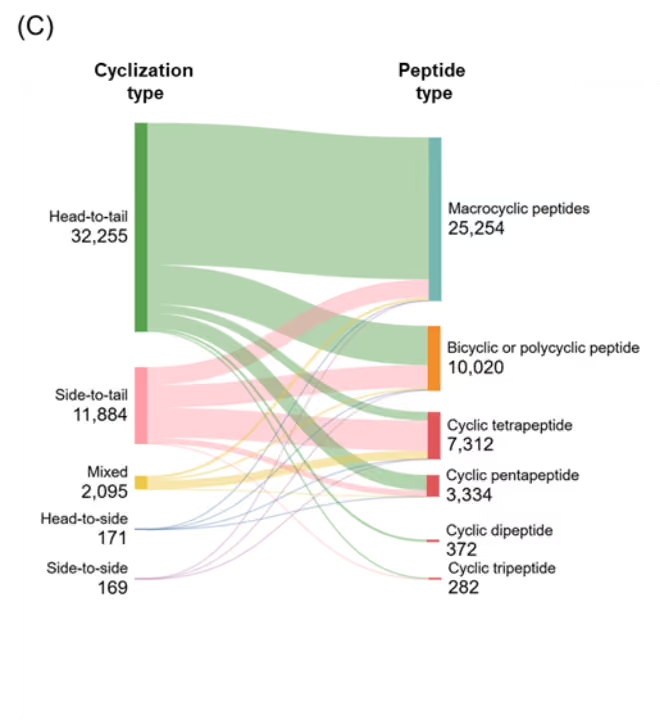
Figure 5: Distribution of identified cyclic peptides based on their (A) type, and (B) cyclization type. (C) Sankey graph showing co-occurrence between peptide type and cyclization type. Only cyclic peptides indexed with CAS roles THU, or PAC, or PKT were included for the analysis for the period 2020-2025. Source: CAS Content Collection.
Our analysis showed that macrocyclic peptides were the most prevalent, accounting for more than half of the identified cyclic peptides, followed by bicyclic and polycyclic structures (see Figure 5A). In contrast, cyclic dipeptides and tripeptides were least common, suggesting that larger peptides are generally preferred for therapeutic applications due to their superior stability and functional versatility.
Among cyclization strategies, head-to-tail cyclization was the most dominant, accounting for nearly two-thirds of the identified cyclic peptides, followed by side-to-tail and mixed cyclizations. Head-to-side and side-to-side account for a small fraction of cyclic peptides indicating that these cyclization types may not be preferred or as explored (see Figure 5B).
Co-occurrence analysis between peptide type and cyclization strategy revealed that most head-to-tail cyclized peptides form macrocyclic architectures, whereas side-to-tail and mixed cyclizations contribute more diversely to bicyclic/polycyclic structures and smaller ring sizes such as tetrapeptides and pentapeptides (see Figure 5C). Overall, these findings underscore the strong association between cyclization strategy and the resulting peptide architecture.
[H3]: Cyclization bonds and chemical/bioconjugate modifications
Cyclic peptides can be stabilized through diverse cyclization bonds such as disulfide, ether, thioether, ester, and thioester linkages, each imparting unique structural and functional properties.
Disulfide bonds, formed between cysteine residues, are among the most common and confer conformational rigidity, though they can be redox-sensitive. Thioether and ether linkages provide enhanced chemical stability compared to disulfides, as they are resistant to reduction and proteolytic cleavage., formed between cysteine residues, are among the most common and confer conformational rigidity, though they can be redox-sensitive. Thioether and ether linkages provide enhanced chemical stability compared to disulfides, as they are resistant to reduction and proteolytic cleavage.
Ester bonds in cyclic peptides enhance protease resistance, solubility, and conformational control, making them useful for improving stability and drug-like properties. Thioester bonds, meanwhile, play a key role in natural biosynthesis and synthetic cyclization by enabling acyl shifts, facilitating efficient macrocyclization, and mimicking biological pathways. Furthermore, amide to ester substitutions in cyclic peptides can also improve their membrane permeability. These cyclization bonds directly influence the therapeutic potential of cyclic peptides by modulating stability, bioavailability, and target affinity.
Chemical or bioconjugate modifications on peptides constitute a fundamental strategy in the rational design of novel peptide entities and the expansion of their functional repertoire. By applying well-established chemical methodologies, it is possible to modulate key physicochemical parameters, including net charge, hydrophobicity, conformational flexibility, amphiphilicity, and sequence composition that collectively govern peptide stability and biological performance, peptide stability, and biological performance.
Such targeted modifications enable researchers to overcome intrinsic limitations of native peptides, thereby improving pharmacokinetic behavior, enhancing biological activity, and broadening therapeutic applicability. Continued advances in modification strategies are driving progress in peptide science, establishing modified peptides as versatile platforms for mechanistic studies and translational applications.
Some important modifications and their significance are summarized in the table below:
Modification
Significance
Methylation
Improves oral bioavailability
Improves protease resistance
Increases membrane permeability
Glycosylation
Improves solubility and stability, improves half-life
Enhances target binding
Reduces immunogenicity
Lipidation
Increases membrane permeability
Improves receptor selectivity and potency
Increases enzymatic stability
Phosphorylation
Impacts peptide conformation and interactions with target proteins
Sulfation
Enhances receptor binding and improves aqueous solubility
PEGylation
Reduces renal clearance
Prolongs circulation half‑life
Table 1: Peptide modifications and their significance. Source: CAS Content Collection.
Our analysis focused on cyclization bonds (disulfide and thioether) and chemical modifications listed in Table 1. Disulfide bonds were the most common, followed by thioether linkages. Among chemical modifications, N-methylation was most prevalent, followed by complex modifications (involving multiple changes) and lipidation. Glycosylation, PEGylation, sulfation, and phosphorylation were comparatively rare, likely because these modifications are more specialized and often used to enhance solubility, bioavailability, or targeting rather than being broadly applied across peptide classes (see Figure 6A).
Co-occurrence analysis between modifications and peptide type revealed that larger peptides, macrocyclic and bicyclic/polycyclic, exhibit diverse and extensive modifications, whereas smaller peptides tend to have fewer modifications (see Figure 6B). This trend suggests that structural complexity provides more opportunities for functional tailoring, which is critical for optimizing therapeutic performance.
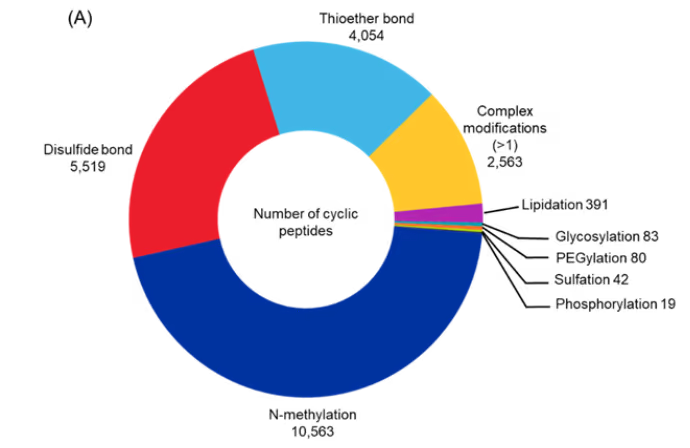
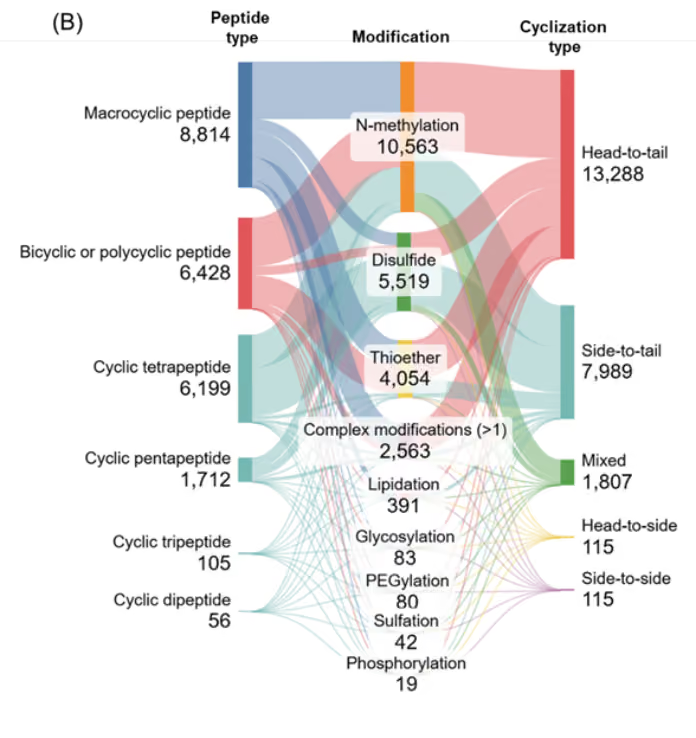
Figure 6: (A) Distribution of cyclic peptides with specified modifications. (B) Sankey graph showing co-occurrence between the various modifications and peptide type and cyclization type. Only cyclic peptides indexed with CAS roles THU, or PAC, or PKT covering the period 2020-2025 were included for the analysis. Source: CAS Content Collection.
We also explored the co-occurences between various peptide types and routes of administration, major therapeutic areas, and potential molecular targets. As seen in Figure 7, this reveals several trends in the probable relationship between peptide design features and preferred administration routes.
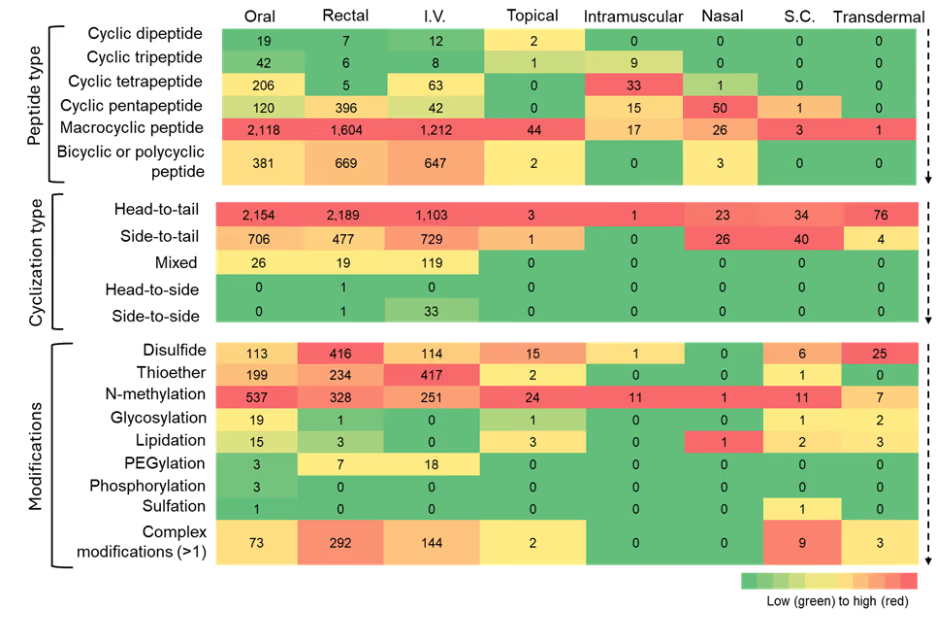
Figure 7: Heatmap summarizing the co-occurrence of cyclic peptide types, cyclization types, and modifications with various administration routes. The heatmap is to be read vertically, with color intensity indicating relative frequency of the number of cyclic peptides. Source: CAS Content Collection.
- Oral, rectal, and intravenous (I.V.) delivery routes dominate almost all peptide classes, indicating that these routes remain the most feasible for diverse peptide structures. Macrocyclic, bicyclic, and polycyclic peptides are heavily represented in oral and I.V. routes, consistent with their enhanced metabolic stability and structural rigidity.
- Subcutaneous (S.C.) and transdermal routes had few cyclic peptides associated with them in our dataset. Macrocyclic and polycyclic peptides are less dominant in intramuscular and nasal routes. Their large size, rigidity, limited aqueous solubility, and poor absorption make them poorly suited for intramuscular and nasal routes, which rely on fast absorption of drugs in systemic circulation.
- Some of the intermediate-sized cyclic peptides (like tetra and pentapeptides) are being explored for intramuscular and nasal routes.
- The topical route appears to have been explored mostly for bacterial skin infections, and inflammatory skin conditions like atopic dermatitis.
- Among cyclization types, head-to-tail, side-to-tail, and mixed type show maximum co-occurrence with oral, rectal, and I.V. routes, likely owing to their high stability and protease resistance.
A closer examination of the modification patterns highlights how different chemical strategies influence compatibility with certain routes of administration.
- N-methylation shows one of the strongest co-occurrences across oral, rectal, and I.V. routes, underscoring its well-known role in increasing protease resistance and improving membrane permeability by reducing hydrogen-bond donors.
- Disulfide and thioether linkages also co-occur prominently across major routes, reflecting their widespread use in stabilizing peptide secondary structures and constraining conformations to enhance metabolic stability.
- Glycosylation and lipidation show distinct distribution, co-occurring mostly with the oral route.
- PEGylation appears to be a modification mostly co-occurring with I.V. formulations, improving their half-life.
- The category of complex modifications, i.e., more than one modification, shows notable co-occurrence with the major routes, reflecting how peptide drug developers are employing combinatorial chemical modification strategies to simultaneously address the multifactorial barriers associated with these routes.
Next, we looked at co-occurrences of cyclic peptides with several diseases and observed a strong prominence of macrocyclic peptides across nearly all therapeutic areas. We found an especially high co-occurrence with cancer, infectious, inflammatory, and autoimmune diseases (see Figure 8).
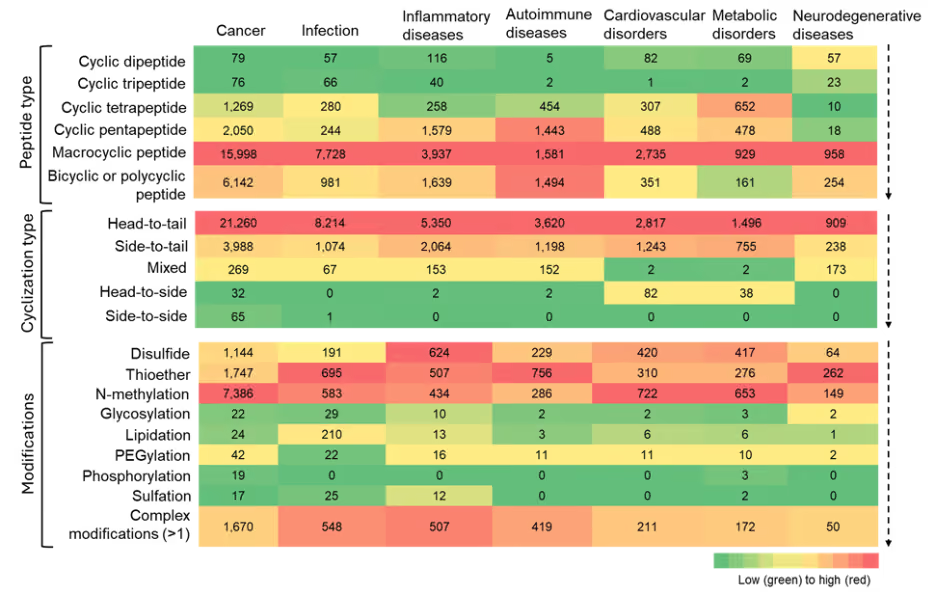
Figure 8: Heatmap summarizing the co-occurrence of cyclic peptide types, cyclization types, and modifications with various therapeutic areas. The heatmap is to be read vertically, with color intensity indicating relative frequency of the number of cyclic peptides. Source: CAS Content Collection.
This trend underscores the structural advantage of macrocycles — enhanced stability, surface area for target engagement, and the ability to modulate traditionally undruggable targets — which aligns well with the mechanistic challenges in these diseases.
Apart from macrocyclic peptides, bicyclic and polycyclic peptides also show relatively high co-occurrence across all therapeutic areas. Cyclic pentapeptides and tetrapeptides show notable co-occurrences, although to a much lesser degree, suggesting that while these scaffolds are valued for their rigidity and selectivity, they have narrower applicability or face greater synthetic constraints.
Our analysis highlights a few specific hotspots like in autoimmune diseases: cyclic pentapeptides, macrocyclic, and bi- or polycyclic peptides appear to be widely explored. Overall, cyclic dipeptides and tripeptides appear to show limited applicability in most therapeutic areas.
Co-occurrence between cyclization patterns and therapeutic areas highlights the well-known preference for head-to-tail cyclized peptides, due to their reliability in imparting conformational restraint and protease resistance without introducing complex chemistries (see Figure 9).
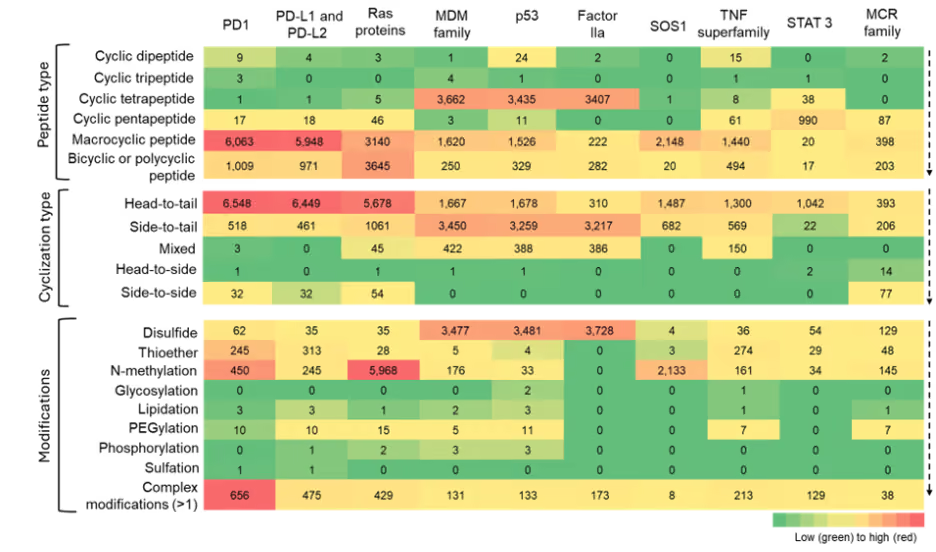
Figure 9: Heatmap summarizing the co-occurence of cyclic peptide types, cyclization types, and modifications across various potential molecular targets. The heatmap is to be read vertically, with color intensity indicating relative frequency of the number of cyclic peptides. Source: CAS Content Collection.
Among modifications, disulfide and thioether linkages, N-methylation, and complex modifications show greater co-occurrences across all therapeutic areas. The relative co-occurrences of other modifications suggest that their use is selective, rather than having broad applicability.
We leveraged our expertly curated CAS concept data to identify proteins that frequently co-occur with our identified cyclic peptides to understand trends relating to different peptide types, cyclization strategies, and chemical modifications. These ten potential molecular targets, shown in Figure 9, represent a diverse collection spanning multiple cellular compartments and functions, yet they share common characteristics that make them attractive for cyclic peptide therapeutics.
The identified targets include cell surface immune regulators (PD1/PDL1/PDL2), intracellular signaling proteins (Ras, SOS1, STAT3), nuclear factors (TP53, MDM family), secreted enzymes (Factor IIa), inflammatory mediators (TNF superfamily), and metabolic receptors (MCR family), with most having significant implications in cancer, autoimmune diseases, or metabolic disorders. While traditional druggability varies widely, from highly druggable targets like Factor IIa and TNF superfamily to historically "undruggable" proteins like Ras and TP53, all represent viable cyclic peptide targets due to their involvement in protein-protein interactions (PPIs), large binding interfaces, or natural peptide ligand recognition.
Their frequent appearance in cyclic peptide literature reflects high therapeutic importance and the unique advantages that cyclic peptides offer: larger binding surfaces for shallow PPI interfaces, enhanced selectivity through conformational constraint, tunable membrane permeability for intracellular targets, and the ability to access binding sites unavailable to traditional small molecules, making them valuable for addressing previously intractable therapeutic targets. It should be noted that not all these publications mention these proteins as direct targets of cyclic peptide therapeutics but may have mentioned them in other contexts.
Figure 9 shows clear patterns: macrocyclic peptides appear to co-occur with several molecular targets, particularly those associated with PD-1/PD-L1, Ras family, and SOS1. This indicates a strong preference for macrocyclic scaffolds in peptide libraries, likely due to their conformational rigidity and stability.
Head-to-tail cyclization emerge as the dominant strategy. Side-to-tail and mixed cyclization approaches appear more often with proteins such as MDM, p53, and Factor IIa, while chemical modifications like N-methylation are often associated with Ras proteins. Disulfide bridges and multi-modifications occur broadly, reflecting design practices aimed at improving stability and permeability.
[H2]: Challenges and opportunities with orally bioavailable cyclic peptides
Traditional biologics, such as monoclonal antibodies and recombinant proteins, have revolutionized the treatment of diseases ranging from cancer to autoimmune disorders. Their ability to engage PPIs with high specificity has enabled the targeting of previously “undruggable” pathways. However, these macromolecules are limited by their need for parenteral administration, which reduces patient compliance, increases healthcare costs, and complicates treatment logistics, especially for chronic conditions requiring frequent dosing.
Conversely, small molecules offer oral bioavailability and ease of administration but often lack the surface area and conformational flexibility needed to effectively modulate PPIs. This dichotomy has created a therapeutic void: targets that demand the precision of biologics but the delivery simplicity of oral drugs remain largely inaccessible. Cyclic peptides, with their intermediate size and tunable properties, are uniquely positioned to fill this gap.
Our analysis of the cyclic peptides dataset revealed distinct trends in the exploration of different drug administration routes across literature. Overall, the oral route dominated the field, accounting for the largest proportion of publications (see Figure 10) and showing a sharp and sustained rise in interest over the past decade. This trend reflects the growing emphasis on improving the oral bioavailability and stability of cyclic peptides, an area historically considered challenging due to their size and polarity. In comparison, other routes, including rectal, I.V., S.C., transdermal, topical, intramuscular, and nasal, appear far less represented with relatively modest and stable publication numbers over time.
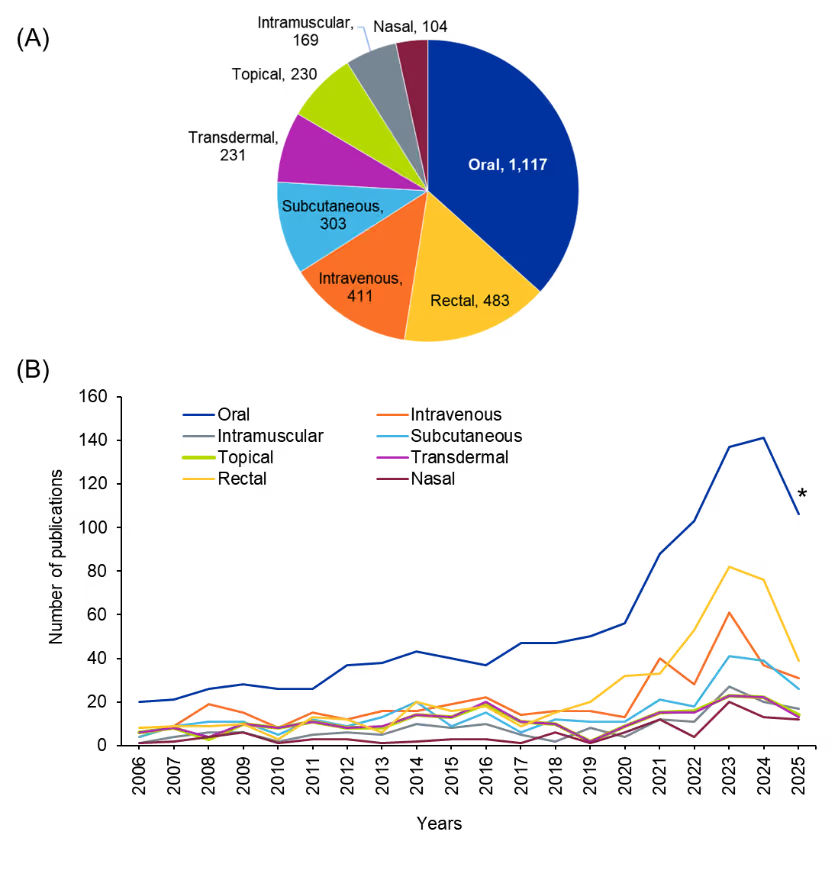
Figure 10: (A) Distribution of publications (journals and patents) and (B) their year-wise trends related to cyclic peptides based on the route of administration. *Data for 2025 is partial through August. Source: CAS Content Collection.
[H3]: Key challenges in oral delivery of cyclic peptides
The poor oral bioavailability of cyclic peptides stems from numerous interdependent factors, including physicochemical constraints, gastrointestinal (GI) tract biology, enzymatic degradation, active efflux, and first-pass metabolism, which result in low and variable oral absorption:
- Physicochemical limitations: Most cyclic peptides possess high molecular weights (typically >500 Da), large polar surface areas, and numerous hydrogen-bond donors and acceptors, which violate conventional oral drug design rules such as Lipinski’s Rule of Five and Veber’s criteria. These properties limit passive membrane permeation, a key requirement for oral absorption.
Cyclization can reduce conformational flexibility and mask polar groups through intramolecular hydrogen bonding, improving proteolytic stability and sometimes permeability. However, residual polarity, size, and excessive rigidity still hinder transcellular diffusion and paracellular passage. Achieving a balance between aqueous solubility (for dissolution in GI fluids) and lipophilicity (for membrane partitioning) remains a major formulation challenge. These physicochemical barriers are fundamental drivers of low permeability observed in in vitro models like Caco-2 cells and in vivo studies.
- Enzymatic degradation and chemical instability: The alimentary canal and the brush border present a rich complement of proteases (pepsin, trypsin, chymotrypsin, brush-border peptidases) and acidic/alkaline microenvironments that cleave peptide bonds or modify labile side chains. While cyclization frequently increases protease resistance relative to linear peptides, many cyclic scaffolds still present susceptible linkages or solvent-exposed residues that enzymes can attack. Moreover, pH-dependent chemical reactions (deamidation, epimerization) can further reduce the fraction of intact drug reaching absorptive surfaces.
- Mucus barrier and epithelial architecture: The mucus gel overlaying the epithelium acts as a barrier by trapping or slowing the diffusion of molecules that interact with mucin, especially large, charged, or hydrophobic peptides, thereby reducing their effective concentration at the epithelial surface. Studies show that cyclic peptides like cyclosporin A can aggregate gel-forming mucins (MUC2, MUC5AC, MUC5B), further impeding their diffusion.
Beyond the mucus layer, tight junctions restrict paracellular transport to small solutes (typically <500 Da), effectively excluding most cyclic peptides. These junctions form a selectively permeable barrier that blocks macromolecules and peptides from passing between epithelial cells. Dedicated uptake transporters for large cyclic scaffolds are rare. As a result, cyclic peptides rely on transcellular passive diffusion or receptor-/lectin-mediated endocytosis, which are often inefficient.
- Active efflux and first-pass metabolism: Many peptide and macrocyclic scaffolds are substrates for efflux pumps such as P-glycoprotein (P-gp) and multidrug resistance proteins (MRPs), which actively transport them back into the intestinal lumen, thereby reducing net uptake and limiting oral bioavailability. Even when peptides are taken up into enterocytes, they may undergo degradation by intracellular and hepatic enzymes, resulting in substantial first-pass metabolic loss and contributing to interindividual variability in systemic exposure.
Cyclosporine A, a classic example of an orally available cyclic peptide, illustrates how formulation strategies (e.g., microemulsions, liposomes, cyclodextrin complexes) and interactions with transporters like P-gp can lead to a wide range of bioavailability across different patient populations and product types.
- Pharmacokinetic variability and clinical implications: The combined effect of dissolution variability, GI transit, fed/fasted state, microbiome interactions, and transporter/enzyme expression results in erratic PK profiles for many orally dosed cyclic peptides. Low absolute bioavailability often necessitates large oral doses or parenteral alternatives, increasing cost and reducing patient convenience.
[H3]: Strategies to overcome challenges in oral delivery
To translate cyclic peptides into practical oral therapeutics, contemporary strategies operate at three complementary levels: (1) molecular engineering to improve intrinsic permeability and stability, (2) formulation and excipient strategies to protect and present peptides to absorption sites, and (3) device- and platform-level innovations that bypass or actively traverse physiological barriers. Integration across levels, “molecule + formulation + device”, defines successful programs.
1. Molecular and chemical design:
a. Backbone modifications and noncanonical residues: Chemical modifications, such as N-methylation, incorporation of D-amino acids, β-amino acids, and peptidomimetic linkers (e.g., azapeptides, amide isosteres), reduce hydrogen bond donors and proteolytic susceptibility, enhancing membrane permeability and metabolic stability. For example, incorporating D-amino acids into somatostatin analogs leads to increased stability and prolonged half-lives. N-methylation has been used to develop peptide drugs like D-amino-8-D-arginine vasopressin (DDAVP, a synthetic form of vasopressin), which exhibits improved stability and antidiuretic activity. Combining N-methylation with head-to-tail cyclization has yielded peptides with improved oral absorption profiles. Our analysis in Figure 7 also shows the importance of these two in the oral route of administration.
b. Lipidation and prodrugs: Lipidation, the chemical attachment of acyl or alkyl lipid chains to peptides, enhances amphipathicity and is widely employed to improve pharmacokinetic and pharmacodynamic properties. A recent study demonstrated the design and synthesis of cyclic lipidated peptides derived from the C-terminal region of Cx43, aimed at inhibiting hemichannel activity and targeting cardiac endothelium. The prodrug strategy involves chemically modifying peptides into inactive forms that are converted back into active drugs post-absorption. This approach improves key properties such as lipophilicity, membrane permeability, and metabolic stability. Common modifications include masking polar groups through esterification, altering amide bonds to evade enzymatic degradation, and attaching cleavable pro-moieties that aid transport and are activated enzymatically or chemically after uptake.
c. Chameleonic design and intramolecular H-bonding: One promising strategy to enhance the oral bioavailability of cyclic peptides involves engineering chameleonic behavior, which is the ability of a molecule to adopt different conformations depending on its environment. In aqueous conditions, such peptides expose polar groups to maintain solubility, while in low-polarity environments like lipid membranes, they fold to form intramolecular hydrogen bonds, effectively masking polar functionalities and facilitating passive diffusion across membranes. Recent molecular dynamics studies have shown that cyclic peptides can transition between open (soluble) and closed (membrane-permeable) conformations at polar/apolar interfaces, such as those found in lipid bilayers. This conformational adaptability is critical for crossing cellular membranes and is increasingly being incorporated into rational design frameworks for orally bioavailable macrocycles.
d. AI and computational prediction: Recent machine-learning tools specifically trained to predict cyclic peptide membrane permeability and chameleonic behavior (e.g., CycPeptMP and other AI models) accelerate the identification of sequences with favorable oral absorption properties, enabling library-scale preselection prior to synthesis. These computational advances are rapidly shortening discovery timelines and focusing medicinal chemistry efforts.
2. Formulation and excipient strategies:
a. Lipid-based systems: Self-emulsifying drug delivery systems (SEDDS) are isotropic mixtures of oil, surfactant/co-surfactant and solvent/co-solvent that spontaneously emulsify when diluted in aqueous fluids. Their renewed potential for oral peptides has emerged from a set of recent studies demonstrating that peptides could dissolve in the oil phase using the principle of hydrophobic ion pairing (HIP) through which peptide lipophilicity could be increased. Lipid-based nanoparticles, including liposomes, solid lipid nanoparticles (SLNs), and nanostructured lipid carriers (NLCs), offer another approach to peptide delivery. They improve solubilization, stabilize apolar cyclic peptides, and promote lymphatic transport, proven in the commercialization of cyclosporine (Neoral®) and continually applied to newer macrocycles.
b. Polymeric nanoparticles and mucoadhesives: Polymeric nanoparticles — commonly made from biodegradable and biocompatible polymers such as poly lactic-co-glycolic acid (PLGA), polylactic acid (PLA), chitosan, polyhydroxyalkanoates (PHAs), and thiolated polymer systems — can encapsulate peptides within their matrix or adsorb them onto their surface. This capability shields them from luminal enzymes, enables controlled release, and increases residence time via mucoadhesion.
Chitosan derivatives can also transiently modulate tight junctions to enhance paracellular flux. Safety and reversibility of permeation modulation are critical concerns. The pre-activated thiolated chitosan nanoparticles loaded with octreotide increased systemic exposure and sustained the hypoglycaemic effect in rats.
c. Enteric and pH-responsive coatings: Enteric polymers shield peptides from gastric acid and release them in the intestine where absorption potential is greater. Smart polymers that release payloads in target intestinal segments further refine exposure windows and reduce premature degradation.
d. Co-formulation with protease inhibitors and permeation enhancers: Co-administration of enzyme inhibitors is used to protect peptides from degradation in the GI tract by temporarily inhibiting digestive enzymes like trypsin and chymotrypsin. Agents such as aprotinin, soybean trypsin inhibitor, and bacitracin have shown enhanced peptide absorption in preclinical studies. However, concerns around safety, interference with normal digestion, and regulatory challenges limit their clinical application.
Permeation enhancers temporarily increase intestinal epithelial permeability, enabling absorption of peptides with poor membrane permeability. They act by opening tight junctions (enhancing paracellular transport), disrupting cell membranes (facilitating transcellular uptake), and inhibiting efflux transporters to boost intracellular drug levels. A notable clinical example is MYCAPSSA® (octreotide), approved by the FDA in June 2020. It uses Transient Permeation Enhancer (TPE®) technology, an enteric-coated capsule containing an oily suspension of octreotide and sodium caprylate, which transiently opens tight junctions to improve absorption.
3. Device- and platform-level innovations:
a. Ingestible microneedle/biologic devices: A new generation of ingestible devices is redefining oral delivery by physically bypassing traditional absorption barriers and enabling direct translocation of biologics across the GI mucosa. The self-orienting millimeter-scale applicator (SOMA) has demonstrated proof-of-concept for gastric mucosal injection of macromolecules, offering a novel route for systemic delivery without penetrating the stomach wall.
Similarly, Rani Therapeutics’ RaniPill™ platform, an ingestible microneedle capsule, has progressed toward clinical evaluation, supported by promising preclinical and early human data. The LUMI device unfolds in the intestine to deliver drug-loaded microneedles, achieving over 10% bioavailability in pigs without tissue injury.
In a recent study in a swine model, liquid-injecting SOMA (L-SOMA) achieved plasma drug levels comparable to subcutaneous injections within 30 min and up to 80% bioavailability in a swine model. A novel self-unfolding, proximity-enabling device demonstrated enhanced oral delivery of macromolecules like insulin and nisin in rats and pigs, achieving up to 12-fold and 4-fold increases in absorption, respectively.
These platforms can achieve systemic exposure levels comparable to parenteral administration for select macromolecules. Their ability to deliver impermeable peptides, despite extensive chemical and formulation optimization, marks a paradigm shift in oral biologic delivery. While further validation is needed for consistent targeting and long-term safety, these technologies represent a transformative step toward overcoming the bioavailability challenges of cyclic peptides.
b. High-velocity and convective delivery capsules: Emerging platforms that employ mechanical or fluidic forces to transiently penetrate or permeate intestinal tissue (e.g., high-velocity jet capsules, expanding structures) are under development and show promise to deliver intact peptide doses to the submucosa, reducing enzymatic exposure.
c. Transporter exploitation and biomimetic conjugation: Conjugating peptides to molecular moieties recognized by endogenous intestinal transporters (bile acids, dipeptide motifs) can enable receptor- or carrier-mediated uptake. This strategy leverages host physiology for active uptake and is being explored in preclinical programs.
Experience shows no single tactic suffices for all cyclic peptides. Rather, integrated solutions, combining rational sequence design (N-methylation, noncanonical residues), predictive AI selection, protective/targeted formulations (lipids, nanoparticles, enteric coatings), and, where necessary, device-based delivery, produce the best chance of attaining clinically meaningful oral bioavailability.
[H2]: The clinical landscape of cyclic peptides
Cyclic peptide therapeutics demonstrate significant clinical relevance with regulatory approvals spanning over seven decades. Early approvals (1940s–1970s) were predominantly antibacterial agents such as bacitracin, polymyxin B, and vancomycin, administered via intravenous or topical routes. Notably, several compounds from this era utilize thioether bonds as key structural elements, including bacitracin, cyclosporine, and romidepsin., which contribute to their conformational stability and biological activity.
Lipidation and glycosylation modifications have been strategically employed in modern antibiotics such as dalbavancin and oritavancin, as well as selected immunomodulators, to improve pharmacokinetic properties including half-life extension and enhanced tissue penetration. Another important and frequent modification is N-methylation which can modify the conformation, hydrogen bonding potential, and lipophilicity of cyclic peptides, enhancing their membrane permeability and oral bioavailability. Several approved drugs like cyclosporin, vancomycin, daptomycin, and romidepsin have methylation modification.
Disulfide bond formation represents a critical structural feature across multiple therapeutic classes, with 23 of the 52 approved compounds containing intramolecular disulfide bridges that provide conformational constraint and proteolytic stability. Our analysis indicates that disulfide bonds continue to be an important feature in recently studied cyclic peptides (see Figure 5). The approved cyclic peptides featuring disulfide bonds include hormone analogs such as oxytocin, vasopressin, and calcitonin derivatives, as well as somatostatin analogs like octreotide and lanreotide. In contrast, thioether bonds are less prevalent but strategically important in specific compound classes, particularly found in romidepsin and cyclosporine.
Recent approvals (2020–2023) include innovative therapeutics for obesity (setmelanotide), myasthenia gravis (zilucoplan), and invasive fungal infections (rezafungin), demonstrating the continued versatility of cyclic peptides in addressing unmet medical needs. Contemporary developments have witnessed a surge in highly engineered peptides targeting complex pathophysiology including autoimmune disorders, oncology, and metabolic syndromes. The molecular weight range has expanded dramatically from approximately 540 g/mol (romidepsin) to >43,000 g/mol (pegcetacoplan), reflecting significant advances in bioconjugation technologies and structure-based drug design.
The therapeutic landscape of cyclic peptides continues to see remarkable growth, and there are many promising candidates currently advancing through clinical development:
[H3]: Oncology indications
- Certepetide (LSTA-1), a cyclic peptide (989.09 g/mol), simultaneously targets αv-integrins and neuropilin-1 (NRP-1), both of which are key mediators of tumor angiogenesis and metastasis. Developed by Certa Therapeutics, it is currently in Phase II trials (NCT05042128) for metastatic pancreatic ductal adenocarcinoma.
- Paluratide (LUNA18), a macrocyclic compound (1,437.68 g/mol) developed by Chugai Pharmaceutical, disrupts RAS–SOS1 interactions to target KRAS-mutant solid tumors, a historically undruggable pathway. It is under Phase I evaluation (NCT05012618) as an oral monotherapy and in combination with cetuximab, positioning it as a potential first-in-class oral RAS-targeted therapy.
- VT1021, a cyclic peptide (638.76 g/mol) developed by Vigeo Therapeutics, targets the CD36/CD47 immune checkpoint axis to counteract tumor immune evasion in glioblastoma. It is currently in Phase III trials (NCT03970447), addressing the critical need for blood-brain barrier-penetrant therapies that modulate glioblastoma's immunosuppressive microenvironment.
- In the realm of antibody drug conjugates, Zelenectide pevedotin (BT8009), a bicyclic peptide-drug conjugate (~4,171 g/mol) developed by Bicycle Therapeutics, targets Nectin-4 expressing tumors by combining the selectivity of cyclic peptides with cytotoxic potency. It is being evaluated in a Phase III trial (NCT06225596) for urothelial cancer and a Phase II trial (NCT04561362) for Nectin-4 expressing advanced malignancies.
[H3]: Non-oncology indications
- AZP-3813 is a cyclic peptide growth hormone receptor antagonist in Phase I development for acromegaly, offering a potential alternative to somatostatin analogs via subcutaneous administration with improved selectivity and reduced side effects.
- Icotrokinra, an oral cyclic peptide (~1,900 g/mol) developed by Johnson & Johnson and Protagonist Therapeutics, targets the IL-23 receptor to treat immune-mediated diseases including psoriasis and ulcerative colitis. It is in Phase III trials under the ICONIC program for moderate-to-severe psoriatic arthritis (NCT06878404) and ulcerative colitis (NCT07196748), offering a convenient oral alternative to injectable biologics.
- MK-0616, a PCSK9-inhibiting cyclic peptide (1,722.09 g/mol) developed by Merck, has reached Phase III trials (NCT06492291) as an oral treatment for hypercholesterolemia. It represents a significant advance in oral peptide delivery, potentially matching the efficacy of injectable PCSK9 inhibitors with the convenience of oral dosing.
- PL8177, a gut-restricted oral cyclic peptide (996.13 g/mol) developed by Palatin Technologies, targets the melanocortin-1 receptor for active ulcerative colitis and is currently in Phase II trials (NCT05466890).
- PL9643, also from Palatin Technologies, is a cyclic peptide targeting multiple melanocortin receptors (MC1R/3R/4R/5R) for dry eye disease via ophthalmic delivery, currently in Phase II trials (NCT05201170).
- Rusfertide (PTG-300), a cyclic peptide (2,441.96 g/mol) developed by Protagonist Therapeutics, targets the hepcidin pathway for polycythemia vera treatment via subcutaneous delivery. It has advanced to Phase III trials (NCT05210790) as a mechanistically novel alternative to traditional phlebotomy-based treatments.
- Solnatide (AP301), a cyclic peptide (1,923.10 g/mol) developed by APEPTICO, targets epithelial sodium channels (ENaC) via inhaled delivery to promote alveolar fluid clearance in acute respiratory distress syndrome (ARDS). It is currently in Phase II trials (NCT03567577), addressing a critical care indication with few effective existing treatments.
These eleven cyclic peptides demonstrate the maturation of peptide therapeutics as a drug class, with successful applications across oncology, inflammatory conditions, hematology, critical care, endocrinology, and cardiovascular medicine. The diversity of targets, delivery systems, and therapeutic applications highlights the versatility of cyclic peptide scaffolds in addressing complex medical challenges.
[H2]: Future directions for cyclic peptides
Cyclic peptides have emerged as a transformative modality in drug development, bridging the gap between small molecules and biologics. Their unique structural features, conformational rigidity, enhanced proteolytic stability, and high target specificity, make them ideal candidates for addressing previously "undruggable" targets. Nevertheless, despite notable successes, many challenges remain and, with them, many exciting opportunities for future research:
- Enhancing oral bioavailability and cell permeability: One of the most critical challenges for cyclic peptide therapeutics remains achieving oral bioavailability and cellular uptake, particularly for intracellular targets. Structural modifications are at the forefront of permeability enhancement. N-methylation of backbone amides has proven effective in reducing polar surface area while maintaining target affinity, as demonstrated by successful oral cyclic peptides like cyclosporine. Incorporation of non-canonical amino acids, including D-amino acids and β-amino acids, not only enhances permeability but also provides proteolytic resistance. Lipidation strategies, exemplified by semaglutide's fatty acid modification, have shown promise in improving both membrane permeability and pharmacokinetic profiles.
Formulation approaches are equally important. Intestinal permeation enhancers such as sodium caprate and SNAC (sodium N-[8-(2-hydroxybenzoyl)amino]caprylate) have enabled oral delivery of peptides previously limited to parenteral administration. Nanoparticle encapsulation systems, including solid lipid nanoparticles and polymeric micelles, protect cyclic peptides from enzymatic degradation while facilitating transcellular transport. Microneedle patches represent an alternative transdermal delivery route that bypasses gastrointestinal barriers entirely. Additionally, designing cyclic peptides for active transport via peptide transporters (PepT1, PepT2) or exploiting transcytosis pathways can significantly improve oral bioavailability. These innovations collectively promise to transition cyclic peptides from injectable formulations to patient-friendly oral therapeutics within the coming decade.
- De novo design and AI-driven discovery: The landscape of cyclic peptide discovery is rapidly evolving from natural product modification to computational de novo design powered by AI. As Heinis and colleagues emphasized, “new powerful techniques based on rational design and in vitro evolution have enabled the de novo development of cyclic peptide ligands to targets for which nature does not offer solutions.” Generative AI models, such as variational autoencoders (VAEs) and generative adversarial networks (GANs), have enabled the design of cyclic peptide sequences with optimized properties. More recently, diffusion models like CycleDesigner and RFpeptides have shown promise in generating novel macrocyclic scaffolds by learning the distribution of successful peptide structures.
Reinforcement learning (RL) frameworks, including CCPep and PepThink-R1, optimize multiple properties simultaneously like balancing affinity, selectivity, stability, and permeability. Specialized platforms such as PepFlow (flow-matching generative model), CycPeptMPNN (graph-based neural network for cyclic peptides), MultiCycPermea (multimodal permeability prediction), and CyclicChamp (energy-based heuristic design), address unique challenges like ring closure compatibility, conformational preferences, and membrane permeability.
Structure prediction tools have also advanced: AlphaFold3 and its adaptations (e.g., AfCycDesign) now model cyclic peptides with non-canonical amino acids and disulfide bonds with atomic-level accuracy. Rosetta, Des3PI, and the cyclicpeptide Python package integrate structural modeling with predictive analytics for early-stage property analysis. Integrated modeling suites and machine learning-guided tools like NCPepFold predict optimal cyclization strategies and ring conformations, accelerating the design-to-validation pipeline.
By combining these AI-driven platforms with automated synthesis and screening, the field is poised to dramatically shorten discovery timelines, transforming cyclic peptide development from years to months, especially for targets previously considered intractable.
- Expanding the druggable proteome: Cyclic peptides are enabling access to protein targets previously considered undruggable, such as transcription factors, scaffolding proteins, and intrinsically disordered proteins. These molecules can engage shallow or dynamic surfaces, induce conformational changes, and disrupt protein–protein interactions (PPIs) critical to disease pathways. Notable successes include cyclic peptides that inhibit the MDM2–p53 interaction, modulate BCL-2 family proteins, and target RAS-effector complexes, once deemed intractable.
Advances in fragment-based and structure-guided design, combined with display technologies like mRNA and phage display, facilitate the discovery of potent cyclic binders. Moreover, the integration of covalent warheads into cyclic frameworks enhances specificity and expands targeting capabilities, positioning cyclic peptides as versatile tools in next-generation drug discovery.
- Novel cyclization and stabilization techniques: Advances in synthetic chemistry are enabling precise control over cyclic peptide architecture and stability, driving innovation in drug design. Orthogonal cyclization strategies, including head-to-tail, side-chain to side-chain (e.g., disulfide bridges, lactam bonds, hydrocarbon staples), and backbone cyclization, enhance conformational rigidity and proteolytic resistance.
Enzymatic methods such as cyanobactin macrocyclases, split-intein systems, and sortase-mediated ligation offer site-specific and traceless cyclization under mild conditions. Click chemistry approaches like CuAAC, SPAAC, and tetrazine-alkene cycloadditions provide bioorthogonal and rapid macrocycle formation. Metal-mediated techniques, including ruthenium-catalyzed ring-closing metathesis and palladium-catalyzed cross-coupling, introduce structural rigidity and enable incorporation of aromatic linkers.
- Conjugation and multifunctional platforms: Cyclic peptides have emerged as versatile scaffolds for multifunctional therapeutic platforms, extending beyond traditional single-agent modalities. Peptide-drug conjugates (PDCs) combine targeting specificity with cytotoxic payloads, using cleavable linkers (e.g., hydrazone, disulfide, cathepsin-sensitive) or non-cleavable linkers to enhance therapeutic indices.
Bispecific formats, including peptidic bispecific antibodies, enable simultaneous engagement of multiple targets, showing promise in immunotherapy and resistance mitigation. Nanoparticle conjugation strategies utilize cyclic peptides like RGD for integrin-targeted delivery, improving biodistribution and reducing off-target effects. In PROTAC applications, cyclic peptides have been used to improve targeting of PROTACs, facilitating targeted protein degradation and expanding therapeutic scope.
Theranostic platforms integrate cyclic peptides with imaging agents (e.g., PET tracers, fluorophores, MRI contrast agents), enabling real-time monitoring of drug distribution and response. These multifunctional strategies position cyclic peptides as central components in next-generation precision therapeutics.
- Emerging therapeutic areas: Cyclic peptides are poised to expand into new domains including neurological diseases, where they can target intracellular PPIs implicated in neurodegeneration. Their potential in metabolic disorders is also growing, with orally bioavailable macrocyclic peptides being explored for conditions like diabetes and obesity. In the realm of rare diseases and gene regulation, cyclic peptides are emerging as modulators of RNA-binding proteins and epigenetic regulators. Furthermore, their structural versatility makes them promising candidates in the fight against antimicrobial resistance, offering novel scaffolds capable of disrupting bacterial virulence pathways without inducing resistance mechanisms.
Cyclic peptides have become an important focus in current drug discovery because they offer a balance between the selectivity of biologics and the stability of small molecules. Recent advances in synthesis, screening technologies, and design strategies have made it easier to generate diverse and more drug-like cyclic peptide structures. This progress is clearly reflected in the growing number of publications and patents in the past few years, indicating steadily increasing interest from academia and industry.
Going forward, the field is likely to expand further as newer tools help address the remaining challenges related to bioavailability, manufacturing, and clinical translation. Overall, cyclic peptides are emerging as a practical and versatile therapeutic modality, with clear potential for continued growth as technologies mature.




